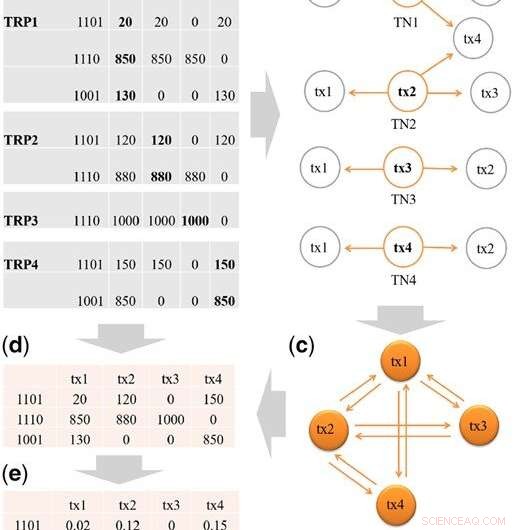
Étapes pour construire la matrice de conception de départ X. (a) TRP de tx1, tx2, tx3 et tx4, et le résumé des modèles d'occupation binaires des TRP. Le transcrit tx5 ne passe pas le filtrage (H = 2,5%) et est filtré de TRP1. Dans chaque motif binaire, le chiffre 1 signifie qu'il y a des lectures provenant d'une eqclass, et 0 sinon. Par exemple, il y a trois eqclasses dans TRP1 :eqclass1, eqclass2 et eqclass3. Pour eq1, le motif binaire est 1101, ce qui signifie trois transcriptions, c'est-à-dire tx1, tx2 et tx4 ont des lectures depuis eq1. (b) Voisins de transcription (TN) pour tx1 à tx4. (c) Illustration de la construction du cluster de transcription (TC) à partir des TN. Nous collectons d'abord les TN de tx1, tx2, tx3 et tx4, puis ajoutez les connexions entre les transcriptions dans le TC. Par exemple, du TN1, on ajoute la connexion de tx1-tx2, tx1-tx3 et tx1-tx4. À la fin, un TC contiendrait toutes les connexions entre les transcrits partageant des exons. (d) L'ensemble unique de motifs binaires est conservé, il reste donc trois modèles uniques : 1101, 1001, 1110. Nous remplissons ensuite le nombre de lectures de chaque TRP source. Par exemple, pour le modèle 1101, dans TRP1, le nombre de lectures est de 20 pour tx1, dans TRP2, le nombre de lectures est de 120 pour tx2 et dans TRP4, le nombre de lectures est de 150 pour tx4. (e) Le nombre total de lectures de chaque transcription en (d) est standardisé pour être égal à 1 pour créer la matrice de conception de départ X. Crédit :DOI :10.1093/bioinformatics/btz640
La technologie omique à haut débit a révolutionné la recherche biologique et biomédicale et de grands volumes de données omiques ont été produits. Pour ça, des outils de calcul pour gérer et analyser les données omics ont été développés et il existe de grands défis dans la façon de traiter et d'interpréter les données omics de la meilleure façon. Wenjiang Deng a travaillé au développement de nouvelles méthodologies statistiques et algorithmes pour l'analyse des données omiques, en utilisant à la fois des données sur le cancer simulées et réelles pour tester les méthodes.
Pouvez-vous décrire certains des résultats de votre thèse ?
Oui, dans ma première étude, nous identifions plusieurs gènes associés à la survie des patients à haut risque de neuroblastome, dit Wenjiang Deng, doctorat étudiant au Département d'épidémiologie médicale et de biostatistique, MEB. Le neuroblastome est le cancer le plus fréquent et le plus mortel chez les jeunes enfants de moins de cinq ans. Nous pensons que nos résultats fourniront des preuves significatives pour le traitement et la gestion des patients. Nos résultats peuvent également être significatifs pour comprendre les mécanismes physiologiques de la maladie.
Comment se fait-il que vous ayez choisi d'étudier ce domaine particulier?
Nous vivons à l'ère du "big data, " et les données de séquençage à haut débit sont les " big data " prédominantes dans les sciences de la vie. Quand j'ai entendu pour la première fois le concept de données omiques, J'ai été étonné par son énorme volume et le grand potentiel de la recherche médicale. De nos jours, il est assez facile de produire des données de séquençage, mais nous avons encore besoin d'outils efficaces et précis pour les analyser, J'ai donc décidé d'étudier le développement d'algorithmes pendant mon doctorat. étudiant.
Que ferez-vous ensuite?
Après ma défense, Je vais rester un moment au MEB pour terminer mes manuscrits. J'irai ensuite à Shenzhen, Chine, et commencer à travailler dans une entreprise de biotechnologie qui vise à développer de nouvelles méthodes de diagnostic précoce des cancers. J'espère que notre travail là-bas contribuera à la santé globale des êtres humains.