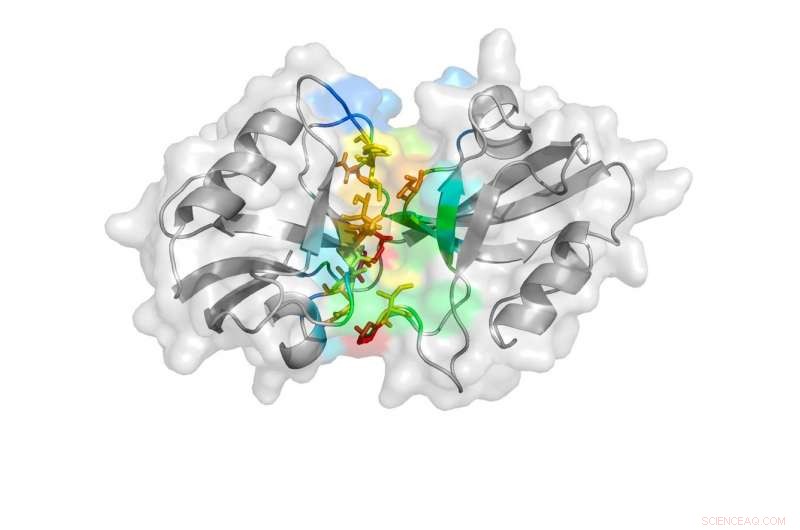
Une représentation dessinée dévoilée par l'étude montre l'état de type fermé des domaines PDZ. Crédit: Communication Nature , doi:10.1038/s41467-018-06133-0
Dans le prolongement d'une recherche publiée il y a un mois dans Méthodes naturelles , une nouvelle approche hybride réalisée par des chercheurs du département de physique et d'astronomie de l'Université Clemson et de l'Université Stony Brook a révélé une structure 3-D d'un fragment de protéine qui pourrait servir de cible médicamenteuse dans le traitement des patients victimes d'un AVC.
La protéine appelée "protéine de densité postsynaptique de 95 kDa (PSD-95)" est positionnée sur les neurones du cerveau qui reçoivent des messages chimiques - des neurotransmetteurs - des neurones adjacents. En recrutant des récepteurs et d'autres protéines auxiliaires, PSD-95 travaille pour maintenir l'intégrité des connexions neuronales au fil du temps, facilitant ainsi la communication neuronale, apprentissage et mémoire.
PSD-95 se compose de cinq parties, ou domaines, que chacun joue un rôle différent dans la fonction globale de la protéine. Deux de ces domaines, appelé PDZ-1 et PDZ-2, ont été montrés pour influencer les symptômes associés à l'AVC ischémique, comme la paralysie ou les troubles de la parole.
"L'une des idées postulées dans la littérature est de créer un médicament multivalent qui cible les deux domaines PDZ car ils sont de nature très similaire. Si vous pouvez empêcher les domaines PDZ de se lier à des protéines ou enzymes particulières, vous pouvez réduire les effets débilitants d'un accident vasculaire cérébral, " a déclaré Hugo Sanabria, auteur principal de l'étude.
Le défi, cependant, est qu'il est presque impossible de concevoir un inhibiteur médicamenteux sans connaître d'abord la structure exacte des domaines PDZ de PSD-95. Ce serait comme conduire à travers le pays sans avoir une carte des États-Unis.
« Les fonctions biologiques des biomolécules sont déterminées par leurs structures, nous avons donc besoin d'informations structurelles et dynamiques détaillées sur les PDZ-1 et -2 pour mieux comprendre leurs rôles fonctionnels et aider à la conception de nouveaux inhibiteurs, " dit Feng Ding, Le collègue de Sanabria ici à Clemson.
Il existe une poignée d'approches pour rendre la structure des biomolécules. Mais dans le cas de PSD-95, chaque approche—spectroscopie RMN, La cristallographie aux rayons X et le transfert d'énergie par résonance de Förster (FRET) ont fourni un modèle structurel différent. Le collaborateur des chercheurs de l'Université Stony Brook, professeur agrégé Mark Bowen au département de physiologie et biophysique, a établi un partenariat avec Sanabria sur ce projet après avoir découvert l'un des modèles structurels incohérents du fragment PSD-95.
Le laboratoire de Sanabria a résolu cette divergence en modélisant d'abord le fragment PSD-95 à l'aide de FRET, une approche qui identifie des configurations possibles de biomolécules. Sous cette méthode, Sanabria a attaché deux molécules photosensibles, appelés chromophores, à deux positions différentes sur le fragment PSD-95. Il a ensuite découvert la distance entre les chromophores en visualisant le fragment au microscope. Cela a été répété plusieurs fois à partir de différents points de fixation.
"Pour l'aspect modélisation, FRET vous donne les distances entre les chromophores, mais cela ne suffit pas à combler toutes les contraintes géométriques de la molécule, donc il faut compter sur autre chose, une autre méthodologie. C'est là que le professeur Ding entre en jeu, ", a déclaré Sanabria.
Ding dirige un laboratoire de biophysique computationnelle à l'Université de Clemson où il utilise un logiciel informatique pour évaluer à quoi ressemblent les biomolécules, bouger et fonctionner. Son approche de la modélisation utilise une simulation informatique connue sous le nom de dynamique moléculaire discrète (DMD) qui cartographie le paysage d'une biomolécule, prédire les trajectoires des protéines lorsqu'elles se replient et interagissent avec d'autres molécules. La simulation suivante peut être lue comme un film, aider les chercheurs à visualiser les comportements des protéines au fil du temps.
"Si vous faites des simulations moléculaires traditionnelles, généralement, vous allez échantillonner une très petite région de l'espace, en particulier pour les molécules plus grosses, vous n'aurez donc pas une bonne vue d'ensemble de l'apparence de la molécule entière, même dans des conditions physiologiques, " Sanabria a déclaré. "La dynamique moléculaire discrète est un moyen beaucoup plus rapide et moins coûteux en calcul d'échantillonner avec précision et rapidement l'espace conformationnel des protéines."
Pour le faire, Sanabria a d'abord obtenu un ensemble de distances en mesurant PSD-95 avec FRET. Dans cette expérience, Sanabria avait 10 échantillons du fragment PSD-95 qui rendaient chacun des distances différentes et trois formes communes - ou conformations - de PSD-95 ont été observées. Encore, sans simulation DMD, il n'y avait aucun moyen pour les chercheurs de savoir quelle distance correspondait à quelle conformation du fragment. Ils saisissent donc chaque distance possible par rapport à chaque forme possible et laissent la simulation faire le reste.
« Une fois que nous avons fait la première simulation, nous avons vu qu'il y avait trois états principaux que PDZ-1 et -2 prenaient. L'un montrait un contact très étroit entre les deux, l'un montrait un ensemble de contacts intermédiaires et l'autre n'avait aucun contact, " dit Ding.
Les chercheurs ont ensuite exécuté à nouveau une simulation DMD sans tenir compte des distances FRET pour confirmer que les trois états observés existent dans la nature et ne sont pas simplement un hasard imposé par les distances FRET. Ils ont en outre sondé les structures en examinant la façon dont les acides aminés individuels, qui constituent les domaines PDZ, lier les uns aux autres. A partir de ces analyses, Ding, Bowen et Sanabria ont pu confirmer que les domaines PDZ prennent deux des trois états observés dans la simulation DMD, celui avec un certain contact et celui sans aucun contact.
"Maintenant, nous avons deux cibles potentielles pour concevoir de nouveaux médicaments qui seront plus efficaces que ceux qui sont actuellement disponibles, " Sanabria a déclaré. "Les perspectives pour les patients victimes d'un AVC sont prometteuses."
Sans dynamique moléculaire discrète, qui peut capturer les changements de conformation qui se produisent à l'échelle de la microseconde, ces deux états auraient été manqués comme ils l'étaient dans les études antérieures.
"La plupart des personnes qui font de la modélisation structurelle guidée par FRET travaillent avec une molécule rigide, comme l'ADN. Si vous avez une molécule rigide, il est facile à modéliser :vous n'avez qu'un seul état à capturer. Vous pouvez assigner les distances FRET et il n'y a vraiment pas de problème, " dit Sanabria. " Dans ce cas, nous avons dépassé cette approche à bien des égards."
Dans les études futures, l'équipe cherche à analyser le potentiel du fragment PSD-95 à s'auto-inhiber en fonction de la propre structure du fragment.