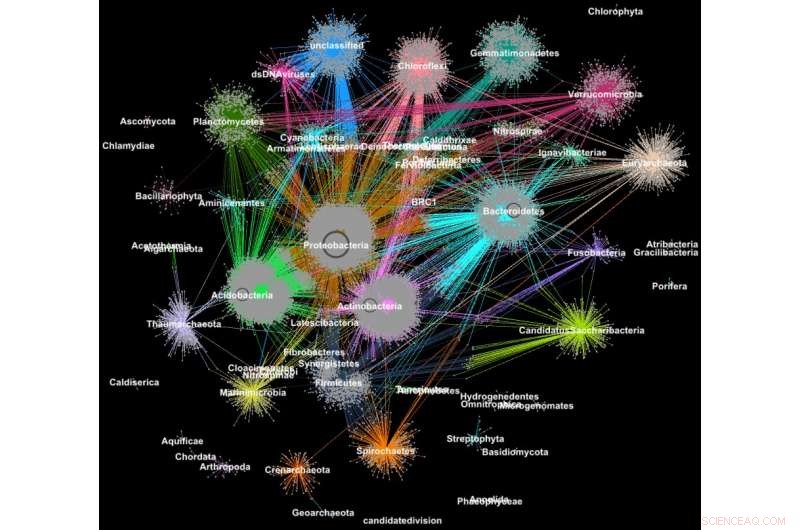
Protéines de métagénomes regroupées en familles selon leur classification taxonomique. Crédit :Georgios Pavlopoulos et Nikos Kyrpides, JGI/Laboratoire de Berkeley
Saviez-vous que les outils utilisés pour analyser les relations entre les utilisateurs de réseaux sociaux ou pour classer les pages Web peuvent également être extrêmement précieux pour donner un sens aux données scientifiques volumineuses ? Sur un réseau social comme Facebook, chaque utilisateur (personne ou organisation) est représenté comme un nœud et les connexions (relations et interactions) entre eux sont appelées arêtes. En analysant ces connexions, les chercheurs peuvent en apprendre beaucoup sur chaque utilisateur :intérêts, passe-temps, habitudes d'achat, copains, etc.
En biologie, des algorithmes de regroupement de graphes similaires peuvent être utilisés pour comprendre les protéines qui exécutent la plupart des fonctions de la vie. On estime que le corps humain à lui seul en contient environ 100, 000 types de protéines différentes, et presque toutes les tâches biologiques—de la digestion à l'immunité—se produisent lorsque ces micro-organismes interagissent les uns avec les autres. Une meilleure compréhension de ces réseaux pourrait aider les chercheurs à déterminer l'efficacité d'un médicament ou à identifier des traitements potentiels pour diverses maladies.
Aujourd'hui, les technologies avancées à haut débit permettent aux chercheurs de capturer des centaines de millions de protéines, gènes et d'autres composants cellulaires à la fois et dans une gamme de conditions environnementales. Des algorithmes de clustering sont ensuite appliqués à ces ensembles de données pour identifier des modèles et des relations qui peuvent indiquer des similitudes structurelles et fonctionnelles. Bien que ces techniques soient largement utilisées depuis plus d'une décennie, ils ne peuvent pas suivre le torrent de données biologiques généré par les séquenceurs et les puces à ADN de nouvelle génération. En réalité, très peu d'algorithmes existants peuvent regrouper un réseau biologique contenant des millions de nœuds (protéines) et de bords (connexions).
C'est pourquoi une équipe de chercheurs du Lawrence Berkeley National Laboratory (Berkeley Lab) et du Joint Genome Institute (JGI) du Department of Energy (DOE) a adopté l'une des approches de clustering les plus populaires de la biologie moderne - l'algorithme Markov Clustering (MCL) - et l'a modifié pour qu'il s'exécute rapidement, efficacement et à grande échelle sur des supercalculateurs à mémoire distribuée. Dans un cas test, leur algorithme haute performance, appelé HipMCL, a réussi un exploit auparavant impossible :regrouper un vaste réseau biologique contenant environ 70 millions de nœuds et 68 milliards d'arêtes en quelques heures, en utilisant environ 140, 000 cœurs de processeur sur le supercalculateur Cori du National Energy Research Scientific Computing Center (NERSC). Un article décrivant ce travail a été récemment publié dans la revue Recherche sur les acides nucléiques .
« Le véritable avantage de HipMCL est sa capacité à regrouper des réseaux biologiques massifs qui étaient impossibles à regrouper avec le logiciel MCL existant, nous permettant ainsi d'identifier et de caractériser le nouvel espace fonctionnel présent dans les communautés microbiennes, " dit Nikos Kyrpides, qui dirige les efforts de science des données sur le microbiome de JGI et le programme Super Prokaryote et est co-auteur de l'article. "De plus, nous pouvons le faire sans sacrifier la sensibilité ou la précision de la méthode d'origine, ce qui est toujours le plus grand défi dans ce genre d'efforts de mise à l'échelle."
« Au fur et à mesure que nos données augmentent, il devient encore plus impératif de déplacer nos outils dans des environnements de calcul haute performance, " ajoute-t-il. " Si vous me demandiez quelle est la taille de l'espace protéique ? La vérité est, nous ne le savons pas vraiment car jusqu'à présent, nous n'avions pas les outils informatiques pour regrouper efficacement toutes nos données génomiques et sonder la matière noire fonctionnelle. »
En plus des progrès de la technologie de collecte de données, les chercheurs choisissent de plus en plus de partager leurs données dans des bases de données communautaires comme le système Integrated Microbial Genomes &Microbiomes (IMG/M), qui a été développé grâce à une collaboration vieille de plusieurs décennies entre des scientifiques de JGI et de la division de recherche informatique (CRD) de Berkeley Lab. Mais en permettant aux utilisateurs de faire des analyses comparatives et d'explorer les capacités fonctionnelles des communautés microbiennes en fonction de leur séquence métagénomique, des outils communautaires comme IMG/M contribuent également à l'explosion des données dans la technologie.
Comment les promenades aléatoires conduisent à des goulots d'étranglement informatiques
Pour maîtriser ce torrent de données, les chercheurs s'appuient sur l'analyse par grappes, ou de regroupement. Il s'agit essentiellement de regrouper des objets afin que les éléments du même groupe (cluster) soient plus similaires que ceux des autres clusters. Depuis plus d'une décennie, les biologistes computationnels ont favorisé le MCL pour regrouper les protéines par similitudes et interactions.
"L'une des raisons pour lesquelles le MCL est populaire parmi les biologistes computationnels est qu'il est relativement sans paramètre ; les utilisateurs n'ont pas à définir une tonne de paramètres pour obtenir des résultats précis et il est remarquablement stable aux petites modifications des données. C'est important car vous devrez peut-être redéfinir une similitude entre les points de données ou vous devrez peut-être corriger une légère erreur de mesure dans vos données. vous ne voulez pas que vos modifications changent l'analyse de 10 clusters à 1, 000 grappes, " dit Aydin Buluç, un scientifique du CRD et l'un des co-auteurs de l'article.
Mais, il ajoute, la communauté de la biologie computationnelle rencontre un goulot d'étranglement informatique car l'outil s'exécute principalement sur un seul nœud informatique, est coûteux en calculs à exécuter et a une grande empreinte mémoire, ce qui limite la quantité de données que cet algorithme peut regrouper.
L'une des étapes les plus gourmandes en calcul et en mémoire de cette analyse est un processus appelé marche aléatoire. Cette technique quantifie la force d'une connexion entre les nœuds, ce qui est utile pour classer et prédire les liens dans un réseau. Dans le cas d'une recherche sur Internet, cela peut vous aider à trouver une chambre d'hôtel pas chère à San Francisco pour les vacances de printemps et même vous indiquer le meilleur moment pour la réserver. En biologie, un tel outil pourrait vous aider à identifier les protéines qui aident votre corps à combattre un virus de la grippe.
Étant donné un graphe ou un réseau arbitraire, il est difficile de connaître le moyen le plus efficace de visiter tous les nœuds et tous les liens. Une marche aléatoire obtient une idée de l'empreinte en explorant l'ensemble du graphique de manière aléatoire ; il commence à un nœud et se déplace arbitrairement le long d'une arête jusqu'à un nœud voisin. Ce processus se poursuit jusqu'à ce que tous les nœuds du réseau de graphes aient été atteints. Étant donné qu'il existe de nombreuses façons différentes de se déplacer entre les nœuds d'un réseau, cette étape se répète de nombreuses fois. Des algorithmes comme MCL continueront à exécuter ce processus de marche aléatoire jusqu'à ce qu'il n'y ait plus de différence significative entre les itérations.
Dans un réseau donné, vous pouvez avoir un nœud connecté à des centaines de nœuds et un autre nœud avec une seule connexion. Les marches aléatoires captureront les nœuds hautement connectés car un chemin différent sera détecté à chaque exécution du processus. Avec ces informations, l'algorithme peut prédire avec un niveau de certitude comment un nœud du réseau est connecté à un autre. Entre chaque course de marche aléatoire, l'algorithme marque sa prédiction pour chaque nœud du graphique dans une colonne d'une matrice de Markov, un peu comme un grand livre, et les clusters finaux sont révélés à la fin. Cela semble assez simple, mais pour les réseaux de protéines avec des millions de nœuds et des milliards d'arêtes, cela peut devenir un problème extrêmement gourmand en calcul et en mémoire. Avec HipMCL, Les informaticiens de Berkeley Lab ont utilisé des outils mathématiques de pointe pour surmonter ces limitations.
« Nous avons notamment conservé intact le backbone MCL, faisant de HipMCL une implémentation massivement parallèle de l'algorithme MCL original, " dit Ariful Azad, informaticien au CRD et auteur principal de l'article.
Bien qu'il y ait eu des tentatives précédentes de paralléliser l'algorithme MCL pour qu'il s'exécute sur un seul GPU, l'outil ne pouvait encore regrouper que des réseaux relativement petits en raison des limitations de mémoire sur un GPU, note Azad.
"Avec HipMCL, nous retravaillons essentiellement les algorithmes MCL pour qu'ils fonctionnent efficacement, en parallèle sur des milliers de processeurs, et le configurer pour tirer parti de la mémoire agrégée disponible dans tous les nœuds de calcul, " ajoute-t-il. " L'évolutivité sans précédent de HipMCL vient de son utilisation d'algorithmes de pointe pour la manipulation de matrices éparses. "
Selon Buluc, effectuer une marche aléatoire simultanément à partir de nombreux nœuds du graphe est mieux calculé en utilisant une multiplication matricielle à matrice creuse, qui est l'une des opérations les plus basiques de la norme GraphBLAS récemment publiée. Buluç et Azad ont développé certains des algorithmes parallèles les plus évolutifs pour la multiplication de matrices creuses de GraphBLAS et modifié l'un de leurs algorithmes de pointe pour HipMCL.
"Le point crucial ici était de trouver le bon équilibre entre parallélisme et consommation de mémoire. HipMCL extrait dynamiquement autant de parallélisme que possible compte tenu de la mémoire disponible qui lui est allouée, " dit Buluc.
HipMCL :regroupement à grande échelle
En plus des innovations mathématiques, un autre avantage de HipMCL est sa capacité à s'exécuter de manière transparente sur n'importe quel système, y compris les ordinateurs portables, postes de travail et grands supercalculateurs. Les chercheurs y sont parvenus en développant leurs outils en C++ et en utilisant les bibliothèques standard MPI et OpenMP.
"Nous avons largement testé HipMCL sur Intel Haswell, Les processeurs Ivy Bridge et Knights Landing au NERSC, en utilisant jusqu'à 2, 000 nœuds et un demi-million de threads sur tous les processeurs, et dans toutes ces courses, HipMCL a réussi à regrouper des réseaux comprenant des milliers à des milliards d'arêtes, " dit Buluç. "Nous voyons qu'il n'y a pas de barrière dans le nombre de processeurs qu'il peut utiliser pour exécuter et constater qu'il peut regrouper les réseaux 1, 000 fois plus rapide que l'algorithme MCL d'origine."
"HipMCL va être vraiment transformationnel pour la biologie computationnelle des mégadonnées, tout comme les systèmes IMG et IMG/M l'ont été pour la génomique du microbiome, " dit Kyrpides. " Cette réalisation témoigne des avantages de la collaboration interdisciplinaire au Berkeley Lab. En tant que biologistes, nous comprenons la science, mais cela a été si précieux de pouvoir collaborer avec des informaticiens qui peuvent nous aider à surmonter nos limites et à nous propulser vers l'avant."
Leur prochaine étape est de continuer à retravailler HipMCL et d'autres outils de biologie computationnelle pour les futurs systèmes exascale, qui sera capable de calculer des quintillions de calculs par seconde. Cela sera essentiel car les données génomiques continuent de croître à un rythme ahurissant, doublant environ tous les cinq à six mois. Cela sera fait dans le cadre du centre de co-conception Exagraph du DOE Exascale Computing Project.