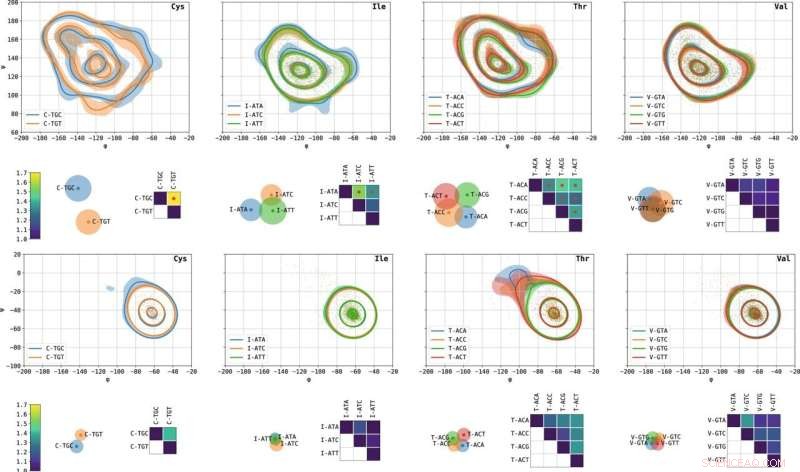
Graphiques Ramachandran spécifiques aux codons d'acides aminés sélectionnés et des distances entre eux. De gauche à droite, la cystéine, l'isoleucine, la thréonine et la valine. Les courbes de niveau représentent les lignes de niveau contenant 10, 50 et 90 % de la masse de probabilité. Les régions ombrées représentent des intervalles de confiance de 10 % à 90 % calculés sur 1 000 bootstraps aléatoires. Les modes β- (haut) et α- (bas) sont représentés. Les matrices montrent L1 distances entre les paires de parcelles de Ramachandran spécifiques aux codons, normalisées de sorte que l'auto-distance soit de 1. Les points rouges indiquent des paires avec des distributions d'angle dièdre significativement différentes en fonction de leur valeur p. Les nuages de points visualisant les matrices de distance ont été obtenus par une variante de mise à l'échelle multidimensionnelle (MDS). Chaque point représente un codon; les distances euclidiennes par paires entre les points se rapprochent de L1 distance entre les codons correspondants. Les cercles se rapprochent des rayons d'incertitude. Plus les deux cercles se chevauchent, moins les tracés de Ramachandran spécifiques aux codons correspondants sont difficiles à distinguer. Crédit :Nature Communications (2022). DOI :10.1038/s41467-022-30390-9
Une étude intégrant des idées biologiques et de nouveaux outils informatiques a découvert de nouvelles associations entre le codage génétique et la structure des protéines, ce qui pourrait potentiellement changer notre façon de penser la production de protéines dans le ribosome, la "chaîne d'assemblage des protéines" de la cellule. La recherche, dirigée par le professeur Alex Bronstein, le Dr Ailie Marx et le Ph.D. l'étudiant Aviv Rosenberg, a été publié dans Nature Communications .
Les protéines, les molécules complexes qui jouent un rôle essentiel dans pratiquement tous les mécanismes biologiques, sont produites par les ribosomes dans un processus appelé traduction. Le ribosome décode les "instructions génétiques" entrantes pour synthétiser les chaînes d'acides aminés - les éléments constitutifs des protéines. Lorsque les acides aminés sont séquentiellement liés ensemble en une longue chaîne, ils se replient en une structure tridimensionnelle unique qui confère à la protéine ses propriétés biologiques et sa fonctionnalité. Des erreurs de traduction peuvent entraîner un mauvais pliage et par la suite des troubles physiologiques, légers ou majeurs.
Les instructions de production de protéines sont transmises au ribosome sous forme de codons, séquences de trois "lettres" du code génétique des nucléotides, qui spécifient l'identité et l'ordre des acides aminés à ajouter par le ribosome à la chaîne protéique. Par exemple, le codon UUU signale l'ajout de l'acide aminé phénylalanine, tandis que le codon UAC demande l'ajout de tyrosine. De cette manière, la séquence de codons code pour la séquence unique d'acides aminés caractéristique de chaque protéine. Cette cartographie des codons génétiques aux acides aminés utilisés dans la traduction est commune à toutes les créatures vivantes de la planète et est considérée comme un mécanisme primitif.
Comme si tout cela n'était pas assez compliqué, il est important de souligner qu'il y a 61 codons qui sont décodés en seulement 20 acides aminés. En d'autres termes, tous les acides aminés sauf deux sont codés par plusieurs codons.
C'est là que la recherche actuelle entre en scène. Sur la base d'expériences menées dans les années 1960 et 1970, le dogme accepté stipule que les protéines ne portent aucune "mémoire" du codon spécifique à partir duquel chaque acide aminé a été traduit tant que l'identité de l'acide aminé reste inchangée. Ces premières expériences sur le repliement des protéines utilisaient des dénaturants chimiques pour déplier les protéines entièrement formées, puis ont démontré que lors de l'élimination de ces produits chimiques, la chaîne protéique pouvait se replier spontanément pour retrouver sa structure et sa fonction d'origine. Ces expériences ont suggéré que seule la séquence d'acides aminés, et non la séquence de codons spécifiques, détermine la structure d'une protéine. Compte tenu de ce dogme, les mutations qui modifient le codage génétique sans modifier l'acide aminé sont largement qualifiées de "silencieuses" et considérées comme sans conséquence pour la structure et la fonction des protéines.
L'équipe de recherche du Technion a découvert une association entre l'identité du codon et la structure locale de la protéine traduite, ce qui suggère que ce n'est peut-être pas le cas général et que les protéines peuvent en effet "se souvenir" des instructions spécifiques à partir desquelles elles ont été synthétisées. L'équipe de recherche a analysé des milliers de structures protéiques tridimensionnelles à l'aide d'outils dédiés qu'elle a développés, qui intègrent des méthodes informatiques avancées, l'apprentissage automatique et les statistiques. De cette façon, ils ont comparé avec précision les distributions des angles formés dans ces structures sous différents codes génétiques synonymes. Leurs résultats montrent que pour certains codons, il existe une dépendance statistique significative entre l'identité du codon et la structure locale de la protéine, à la position de l'acide aminé codé par ce codon.
Les chercheurs soulignent que les résultats ne permettent toujours pas de faire la lumière sur la direction de la relation causale, ce qui signifie qu'il n'est pas encore possible de dire si un changement dans le codage génétique peut provoquer un changement dans la structure protéique locale ou si des changements structurels peuvent provoquer codage différent, par exemple par des processus évolutifs. Cette question est à la base d'une étude de recherche ultérieure actuellement menée par le groupe. According to Dr. Marx, a biologist by training and education, "If we find in subsequent research that the codon indeed has a causal effect on protein folding, this is likely to have a huge impact on our understanding of protein folding, as well as on future applications, such as engineering new proteins."
Dr. Marx emphasizes that the discovery presented in the article would not have been possible without Prof. Bronstein's computer and analysis skills. "This research is truly interdisciplinary, because biology alone cannot cope with such vast quantities of data without the help of data science, and computer scientists cannot themselves perform research of this kind, since they lack familiarity with the complex biological processes being probed. Therefore, our research highlights the huge advantage of interdisciplinary research that integrates skills from different fields to create a whole that is greater than the sum of its parts."














