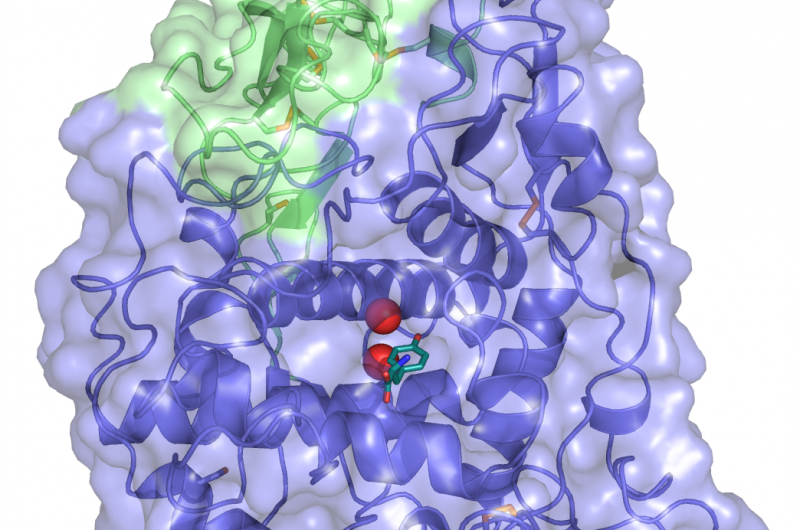
En utilisant une approche de modélisation informatique qu'ils ont développée, Les biologistes du MIT ont identifié trois protéines différentes qui peuvent se lier sélectivement à chacune des trois cibles similaires, tous les membres de la famille de protéines Bcl-2. Crédit :Vincent Xue
Concevoir des protéines synthétiques pouvant agir comme médicaments contre le cancer ou d'autres maladies peut être un processus fastidieux :il s'agit généralement de créer une bibliothèque de millions de protéines, puis cribler la bibliothèque pour trouver des protéines qui se lient à la bonne cible.
Les biologistes du MIT ont maintenant mis au point une approche plus raffinée dans laquelle ils utilisent la modélisation informatique pour prédire comment différentes séquences de protéines interagiront avec la cible. Cette stratégie génère un plus grand nombre de candidats et offre également un meilleur contrôle sur une variété de caractères protéiques, dit Amy Keating, professeur de biologie et de génie biologique et chef de l'équipe de recherche.
« Notre méthode vous offre un terrain de jeu beaucoup plus vaste où vous pouvez sélectionner des solutions très différentes les unes des autres et qui auront des forces et des passifs différents, " dit-elle. " Notre espoir est que nous pouvons fournir un plus large éventail de solutions possibles pour augmenter le débit de ces premiers hits en utiles, molécules fonctionnelles."
Dans un article paru dans le Actes de l'Académie nationale des sciences la semaine du 15 octobre, Keating et ses collègues ont utilisé cette approche pour générer plusieurs peptides pouvant cibler différents membres d'une famille de protéines appelée Bcl-2, qui aident à stimuler la croissance du cancer.
Les récents récipiendaires de doctorat Justin Jenson et Vincent Xue sont les principaux auteurs de l'article. Les autres auteurs sont le postdoctorant Tirtha Mandal, ancienne technicienne de laboratoire Lindsey Stretz, et ancien post-doctorant Lothar Reich.
Modélisation des interactions
Médicaments protéinés, aussi appelé biopharmaceutique, sont une classe de médicaments en croissance rapide qui sont prometteuses pour le traitement d'un large éventail de maladies. La méthode habituelle pour identifier de tels médicaments consiste à cribler des millions de protéines, soit choisis au hasard, soit sélectionnés en créant des variantes de séquences protéiques qui se sont déjà révélées être des candidats prometteurs. Cela implique de concevoir des virus ou des levures pour produire chacune des protéines, puis les exposer à la cible pour voir lesquels se lient le mieux.
"C'est l'approche standard :soit de manière totalement aléatoire, ou avec des connaissances préalables, concevoir une bibliothèque de protéines, puis aller pêcher dans la bibliothèque pour en sortir les membres les plus prometteurs, ", dit Keating.
Bien que cette méthode fonctionne bien, il produit généralement des protéines qui sont optimisées pour un seul trait :à quel point il se lie à la cible. Il ne permet aucun contrôle sur d'autres fonctionnalités qui pourraient être utiles, tels que des traits qui contribuent à la capacité d'une protéine à pénétrer dans les cellules ou à sa tendance à provoquer une réponse immunitaire.
"Il n'y a pas de moyen évident de faire ce genre de chose - spécifier un peptide chargé positivement, par exemple, en utilisant le criblage de bibliothèque par force brute, ", dit Keating.
Une autre caractéristique souhaitable est la capacité d'identifier des protéines qui se lient étroitement à leur cible mais pas à des cibles similaires, ce qui permet de s'assurer que les médicaments n'ont pas d'effets secondaires imprévus. L'approche standard permet aux chercheurs de le faire, mais les expériences deviennent plus lourdes, dit Keating.
La nouvelle stratégie implique d'abord la création d'un modèle informatique qui peut relier les séquences peptidiques à leur affinité de liaison pour la protéine cible. Pour créer ce modèle, les chercheurs ont d'abord choisi environ 10, 000 peptides, chacun de 23 acides aminés de longueur et de structure hélicoïdale, et testé leur liaison à trois membres différents de la famille Bcl-2. Ils ont intentionnellement choisi des séquences dont ils savaient déjà qu'elles se lieraient bien, plus d'autres qu'ils savaient pas, le modèle pourrait donc incorporer des données sur une gamme de capacités de liaison.
A partir de cet ensemble de données, le modèle peut produire un « paysage » de la façon dont chaque séquence peptidique interagit avec chaque cible. Les chercheurs peuvent ensuite utiliser le modèle pour prédire comment d'autres séquences vont interagir avec les cibles, et générer des peptides qui répondent aux critères souhaités.
En utilisant ce modèle, les chercheurs ont produit 36 peptides censés lier étroitement un membre de la famille mais pas les deux autres. Tous les candidats ont obtenu de très bons résultats lorsque les chercheurs les ont testés expérimentalement, ils ont donc essayé un problème plus difficile :identifier des protéines qui se lient à deux des membres mais pas au troisième. Beaucoup de ces protéines ont également été couronnées de succès.
"Cette approche représente un changement de poser un problème très spécifique et ensuite concevoir une expérience pour le résoudre, investir un peu de travail en amont pour générer ce paysage de la façon dont la séquence est liée à la fonction, capturer le paysage dans un modèle, et pouvoir ensuite l'explorer à volonté pour de multiples propriétés, ", dit Keating.
Sagar Kharé, professeur agrégé de chimie et de biologie chimique à l'Université Rutgers, affirme que la nouvelle approche est impressionnante dans sa capacité à discriminer entre des cibles protéiques étroitement liées.
« La sélectivité des médicaments est essentielle pour minimiser les effets hors cible, et souvent la sélectivité est très difficile à coder car il y a tellement de concurrents moléculaires d'apparence similaire qui lieront également le médicament en dehors de la cible visée. Ce travail montre comment coder cette sélectivité dans le design lui-même, " dit Khare, qui n'a pas participé à la recherche. "Des applications dans le développement de peptides thérapeutiques s'ensuivront presque certainement."
Médicaments sélectifs
Les membres de la famille des protéines Bcl-2 jouent un rôle important dans la régulation de la mort cellulaire programmée. La dérégulation de ces protéines peut inhiber la mort cellulaire, aider les tumeurs à se développer sans contrôle, tant de sociétés pharmaceutiques ont travaillé sur le développement de médicaments qui ciblent cette famille de protéines. Pour que ces médicaments soient efficaces, il peut être important pour eux de cibler une seule des protéines, car les perturber tous pourrait provoquer des effets secondaires nocifs dans les cellules saines.
"Dans de nombreux cas, les cellules cancéreuses semblent n'utiliser qu'un ou deux membres de la famille pour favoriser la survie cellulaire, " Keating dit. " En général, il est reconnu qu'avoir un panel d'agents sélectifs serait bien mieux qu'un outil grossier qui les assommerait tous."
Les chercheurs ont déposé des brevets sur les peptides qu'ils ont identifiés dans cette étude, et ils espèrent qu'ils seront davantage testés en tant que médicaments possibles. Le laboratoire de Keating travaille maintenant à l'application de cette nouvelle approche de modélisation à d'autres cibles protéiques. Ce type de modélisation pourrait être utile non seulement pour développer des médicaments potentiels, mais aussi générer des protéines pour une utilisation dans des applications agricoles ou énergétiques, elle dit.
Cette histoire est republiée avec l'aimable autorisation de MIT News (web.mit.edu/newsoffice/), un site populaire qui couvre l'actualité de la recherche du MIT, innovation et enseignement.