Crédit :Université du Michigan
Dans une conclusion qui pourrait aider à ouvrir la voie à des carburants plus propres et à une industrie chimique plus durable, des chercheurs de l'Université du Michigan ont utilisé l'apprentissage automatique pour prédire comment les compositions d'alliages métalliques et d'oxydes métalliques affectent leurs structures électroniques.
La structure électronique est la clé pour comprendre comment le matériau se comportera en tant que médiateur, ou catalyseur, de réactions chimiques.
« Nous apprenons à identifier les empreintes digitales des matériaux et à les connecter aux performances du matériau, " a déclaré Bryan Goldsmith, le professeur assistant Dow Corning en génie chimique.
Une meilleure capacité à prédire quelles compositions de métaux et d'oxydes métalliques sont les meilleures pour guider quelles réactions pourraient améliorer les processus chimiques à grande échelle tels que la production d'hydrogène, production d'autres carburants et engrais, et la fabrication de produits chimiques ménagers tels que le savon à vaisselle.
« L'objectif de nos recherches est de développer des modèles prédictifs qui relieront la géométrie d'un catalyseur à ses performances. De tels modèles sont au cœur de la conception de nouveaux catalyseurs pour des transformations chimiques critiques, " dit Suljo Linic, le professeur Martin Lewis Perl Collegiate de génie chimique.
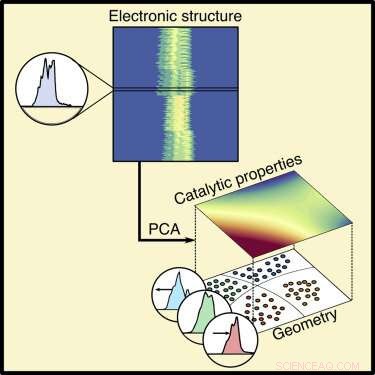
L'une des principales approches pour prédire comment un matériau se comportera en tant que médiateur potentiel d'une réaction chimique consiste à analyser sa structure électronique, en particulier la densité d'états. Cela décrit le nombre d'états quantiques disponibles pour les électrons dans les molécules en réaction et les énergies de ces états.
D'habitude, la densité électronique des états est décrite avec des statistiques récapitulatives - une énergie moyenne ou un biais qui révèle si davantage d'états électroniques sont au-dessus ou en dessous de la moyenne, etc.
"C'est bon, mais ce ne sont que de simples statistiques. Vous pourriez manquer quelque chose. Avec l'analyse en composantes principales, vous prenez juste tout et trouvez ce qui est important. Vous ne jetez pas simplement des informations, " dit Goldsmith.
L'analyse en composantes principales est une méthode classique d'apprentissage automatique, enseigné dans les cours d'introduction à la science des données. Ils ont utilisé la densité électronique d'états comme entrée pour le modèle, comme la densité d'états est un bon prédicteur de la façon dont la surface d'un catalyseur va s'adsorber, ou en lien avec, atomes et molécules qui servent de réactifs. Le modèle lie la densité d'états à la composition du matériau.
Contrairement à l'apprentissage automatique conventionnel, qui est essentiellement une boîte noire qui entre des données et offre des prédictions en retour, l'équipe a créé un algorithme qu'ils pouvaient comprendre.
"Nous pouvons voir systématiquement ce qui change dans la densité d'états et corréler cela avec les propriétés géométriques du matériau, " dit Jacques Esterhuizen, doctorant en génie chimique et premier auteur de l'article en Catalyse chimique .
Ces informations aident les ingénieurs chimistes à concevoir des alliages métalliques pour obtenir la densité d'états qu'ils souhaitent pour la médiation d'une réaction chimique. Le modèle reflétait avec précision les corrélations déjà observées entre la composition d'un matériau et sa densité d'états, ainsi que de découvrir de nouvelles tendances potentielles à explorer.
Le modèle simplifie la densité d'états en deux parties, ou composants principaux. Une pièce couvre essentiellement la façon dont les atomes du métal s'emboîtent. Dans un alliage métallique en couches, cela inclut si le métal souterrain sépare les atomes de surface ou les serre ensemble, et le nombre d'électrons que le métal souterrain contribue à la liaison. L'autre élément n'est que le nombre d'électrons que les atomes métalliques de surface peuvent contribuer à la liaison. A partir de ces deux composantes principales, ils peuvent reconstituer la densité d'états dans le matériau.
Ce concept fonctionne également pour la réactivité des oxydes métalliques. Dans ce cas, la préoccupation est la capacité de l'oxygène à interagir avec les atomes et les molécules, qui est lié à la stabilité de l'oxygène de surface. Les oxygènes de surface stables sont moins susceptibles de réagir, alors que les oxygènes de surface instables sont plus réactifs. Le modèle a capturé avec précision la stabilité de l'oxygène dans les oxydes métalliques et les pérovskites, une classe d'oxydes métalliques.