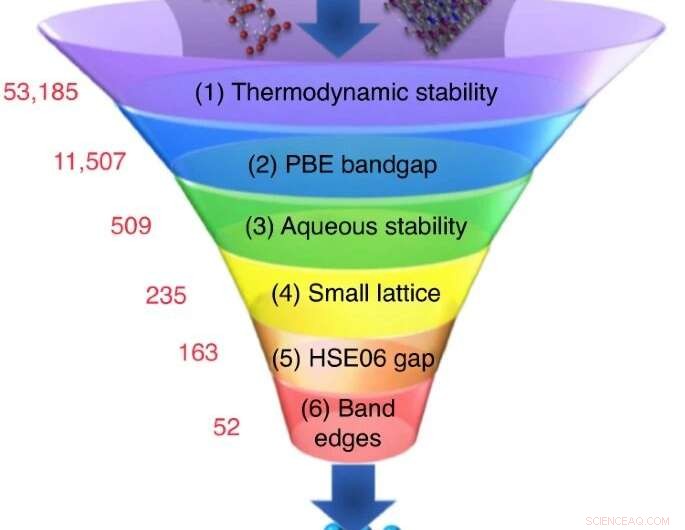
Un grand nombre de matériaux candidats sont choisis dans des bases de données expérimentales ou computationnelles, et une séquence de calculs de sélection réduit leur nombre à un petit ensemble de candidats possédant les propriétés les plus prometteuses. Crédit :Nicola Marzari
Nicola Marzari, responsable du laboratoire Théorie et Simulation des Matériaux à l'EFPL et directeur du PRN MARVEL, vient de publier une revue des méthodes de structure électronique dans le cadre d'une édition spéciale Insight on Computational Materials Design, publié par Matériaux naturels . L'article, écrit avec Andrea Ferretti du CNR-Instituto Nanoscienze et Chris Wolverton de la Northwestern University, donne un aperçu de ces méthodes, discute de leur application à la prédiction des propriétés des matériaux, et examine différentes stratégies utilisées pour cibler les objectifs plus larges de la conception et de la découverte de matériaux. Regarder vers l'avant, les auteurs considèrent les défis émergents dans l'exactitude prédictive des calculs, et en abordant la complexité réelle des matériaux et des dispositifs. Ils soulignent également l'importance des infrastructures informatiques qui soutiennent une telle recherche, et comment la planification de leur financement et des modèles de carrière qui les soutiennent ne fait que commencer à émerger.
Au cours des 20 dernières années, les simulations de premiers principes sont devenues puissantes, outils largement utilisés dans de nombreux, divers domaines de la science et de l'ingénierie. De la nanotechnologie à la science planétaire, de la métallurgie aux matériaux quantiques, ils ont accéléré l'identification, caractérisation, et l'optimisation des matériaux énormément. Ils ont conduit à des prédictions étonnantes - du transport thermique ultrarapide à la supraconductivité à médiation électron-phonon dans les hydrures à l'émergence de bandes plates dans le graphène à bicouche torsadée - qui ont inspiré des expériences remarquables.
La poussée actuelle pour compléter les expériences par des simulations ; a continué, croissance rapide de la capacité de débit informatique; la capacité de l'apprentissage automatique et de l'intelligence artificielle à accélérer la découverte de matériaux ainsi que la promesse d'accélérateurs perturbateurs tels que l'informatique quantique pour des tâches exponentiellement coûteuses signifient qu'il est évident que ces méthodes deviendront de plus en plus pertinentes au fil du temps. Le moment est alors venu de passer en revue les capacités ainsi que les limites des méthodes de structure électronique sous-jacentes à ces simulations. Marzari, Ferretti et Wolverton abordent cette tâche dans l'article « Méthodes de structure électronique pour la conception de matériaux, " vient de paraître dans Matériaux naturels .
"Les simulations n'échouent pas de manière spectaculaire, mais peuvent subtilement passer d'inestimables à à peine assez bonnes à tout simplement inutiles, " les auteurs ont déclaré dans le journal. " Les raisons de l'échec sont multiples, de l'extension des capacités des méthodes à l'abandon de la complexité des matériaux réels. Mais les simulations sont également irremplaçables :elles peuvent évaluer des matériaux dans des conditions de pression et de température si extrêmes qu'aucune expérience sur terre n'est capable de les reproduire, ils peuvent explorer avec une agilité toujours croissante le vaste espace des phases et des compositions des matériaux à la recherche de cette percée insaisissable des matériaux, et ils peuvent identifier directement les causes microscopiques et l'origine d'une propriété macroscopique. Durer, ils partagent avec toutes les branches de la science informatique un élément clé de la recherche :ils peuvent être rendus reproductibles, ouverts et partageables d'une manière qu'aucune infrastructure physique ne le sera jamais. »
Les auteurs examinent d'abord le cadre de la théorie de la fonctionnelle de la densité (DFT) et donnent un aperçu des approches de plus en plus complexes qui peuvent améliorer la précision ou étendre la portée des simulations. Ils discutent ensuite des capacités développées par la science informatique des matériaux pour exploiter cette boîte à outils et fournir des prédictions sur les propriétés des matériaux dans des conditions réalistes d'une complexité toujours croissante. Finalement, ils mettent en évidence comment les approches basées sur la physique ou les données peuvent fournir des haut débit, ou des voies d'intelligence artificielle pour la découverte de matériaux, et expliquer comment ces efforts changent l'ensemble de l'écosystème de la recherche.
Regarder vers l'avant, les auteurs disent que développer des méthodes qui peuvent évaluer la stabilité thermodynamique, conditions de synthèse, manufacturabilité, et la tolérance des propriétés prédites aux défauts intrinsèques et extrinsèques dans les nouveaux matériaux sera un défi important. Les chercheurs peuvent avoir besoin d'augmenter les estimations DFT par des méthodes de structure électronique plus avancées ou des algorithmes d'apprentissage automatique pour améliorer la précision, et utiliser des méthodes de calcul pour traiter des conditions réalistes telles que les entropies vibrationnelles, la concentration des défauts et les potentiels électrochimiques appliqués.
Finalement, étant donné le rôle accru que de telles méthodes sont susceptibles de jouer dans les décennies à venir, les auteurs notent que le soutien et la planification des infrastructures de calcul nécessaires - des logiciels scientifiques largement utilisés, la vérification des codes et la validation des théories, la diffusion et la conservation des données informatiques, les outils et les flux de travail ainsi que les modèles de carrière associés qu'ils impliquent et nécessitent, commencent tout juste à émerger.