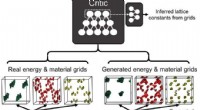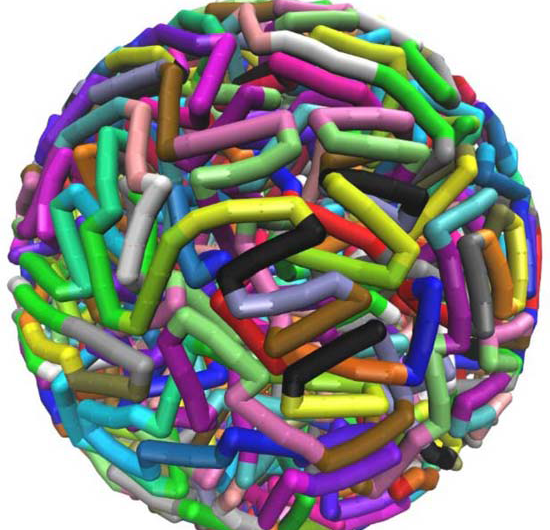
Spécifique à la séquence, induit par torsion, configurations élastiques coudées, générés par des simulations de dynamique moléculaire sur des supercalculateurs du Texas Advanced Computing Center, aider à expliquer combien de longs brins d'ADN peuvent tenir dans de petits espaces. Crédit :Christopher G. Myers, B. Montgomery Pettitt, Branche médicale de l'Université du Texas
Un mystère biologique est au centre de chacune de nos cellules, à savoir :comment un mètre d'ADN peut être ajouté dans l'espace d'un micron (ou un millionième de mètre) à l'intérieur de chaque noyau de notre corps.
Les noyaux des cellules humaines ne sont même pas l'endroit biologique le plus peuplé que nous connaissions. Certains bactériophages, des virus qui infectent et se répliquent au sein d'une bactérie, ont un ADN encore plus concentré.
"Comment ça entre là-dedans ?" B. Montgomery (Monte) Pettitt, biochimiste et professeur à la branche médicale de l'Université du Texas, demande. "C'est un polymère chargé. Comment surmonte-t-il la répulsion à sa densité cristalline liquide ? Combien d'ordre et de désordre sont autorisés, et comment cela joue-t-il un rôle dans les acides nucléiques ?"
En utilisant les supercalculateurs Stampede et Lonestar5 de l'Université du Texas au Texas Advanced Computing Center (TACC) d'Austin, Pettitt étudie comment l'ADN des phages se replie dans des espaces hyper-confinés.
Écrivant dans le numéro de juin 2017 du Journal de chimie computationnelle , il a expliqué comment l'ADN peut surmonter à la fois la répulsion électrostatique et sa rigidité naturelle.
La clé pour y parvenir ? Kinks.
L'introduction de torsions ou de courbes nettes dans des configurations d'ADN emballé dans une enveloppe sphérique réduit considérablement les énergies et les pressions globales de la molécule, selon Pettitt.
Lui et ses collaborateurs ont utilisé un modèle qui déforme et plie l'ADN toutes les 24 paires de bases, qui est proche de la longueur moyenne qui est prédite à partir de la séquence d'ADN du phage. L'introduction de tels défauts persistants réduit non seulement l'énergie de flexion totale de l'ADN confiné, mais réduit également la composante électrostatique de l'énergie et de la pression.
"Nous montrons qu'un large ensemble de configurations de polymères est cohérent avec les données structurelles, " lui et son collaborateur Christopher Myers, également de la branche médicale de l'Université du Texas, a écrit.
De telles informations ne peuvent pas être obtenues uniquement en laboratoire. Ils nécessitent des superordinateurs qui servent de microscopes moléculaires, cartographier le mouvement des atomes et des liaisons atomiques à des échelles de longueur et de temps qu'il n'est pas possible d'étudier avec des expériences physiques seules.
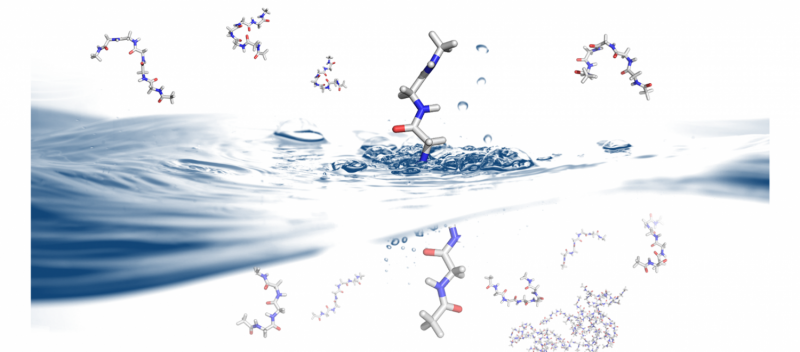
Comment et pourquoi les protéines se replient est un problème qui a des implications pour la conception et la thérapeutique des protéines. B. Montgomery Pettitt et son groupe de recherche de la branche médicale de l'Université du Texas utilisent les superordinateurs Stampede et Lonestar5 du Texas Advanced Computing Center pour explorer la dynamique du repliement des protéines en solution. Crédit :Christopher G. Myers, B. Montgomery Pettitt, Branche médicale de l'Université du Texas
« Dans le domaine de la biologie moléculaire, il y a une merveilleuse interaction entre la théorie, expérimentation et simulation, " Pettitt a déclaré. "Nous prenons des paramètres d'expériences et voyons s'ils sont en accord avec les simulations et les théories. Cela devient la méthode scientifique pour la façon dont nous avançons maintenant nos hypothèses. »
Des problèmes comme ceux qui intéressent Pettitt ne peuvent pas être résolus sur un ordinateur de bureau ou un cluster de campus typique, mais nécessitent des centaines de processeurs informatiques travaillant en parallèle pour imiter les mouvements infimes et les forces physiques des molécules dans une cellule.
Pettitt est en mesure d'accéder aux superordinateurs de TACC en partie grâce à un programme unique connu sous le nom de Journal de chimie computationnelle initiative, ce qui rend les ressources de calcul de TACC, l'expertise et la formation disponibles pour les chercheurs au sein des 14 institutions de l'Université du Texas Systems.
"La recherche informatique, comme celle du Dr Pettitt, qui cherche à combler notre compréhension de la physique, chimique, et finalement des phénomènes biologiques, implique tellement de calculs qu'il n'est vraiment accessible que sur de gros supercalculateurs comme les systèmes Stampede ou Lonestar5 de TACC, " a déclaré Brian Beck, chercheur en sciences de la vie à la TACC.
« La disponibilité des ressources de supercalcul TACC est essentielle pour ce style de recherche, " dit Pettit.
TROUVER L'ORDRE DANS LES PROTÉINES DÉSORDONNEES
Un autre phénomène qui intéresse depuis longtemps Pettitt est le comportement des protéines intrinsèquement désordonnées (IDP) et des domaines intrinsèquement désordonnés, où les parties d'une protéine ont une forme désordonnée.
Contrairement aux cristaux ou à l'ADN hautement concentré des virus, qui ont distinct, formes rigides, Les personnes déplacées "se replient dans un désordre gluant, " selon Pettitt. Et pourtant, ils sont essentiels pour toutes les formes de vie.
On pense que chez les eucaryotes (organismes dont les cellules ont des sous-structures complexes comme les noyaux), environ 30 pour cent des protéines ont un domaine intrinsèquement désordonné. Plus de 60 % des protéines impliquées dans la signalisation cellulaire (processus moléculaires qui prennent des signaux de l'extérieur de la cellule ou à travers les cellules qui indiquent à la cellule quels comportements activer et désactiver en réponse) ont des domaines désordonnés. De la même manière, 80% des protéines de signalisation liées au cancer ont des régions IDP, ce qui en fait des molécules importantes à comprendre.
Parmi les PDI que Pettitt et son groupe étudient, il y a des facteurs de transcription nucléaires. Ces molécules contrôlent l'expression des gènes et possèdent un domaine de signalisation riche en acide aminé flexible, glycine.

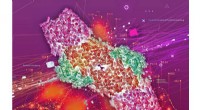
Les images ci-dessus montrent les distributions de densité moyenne sur 21 configurations d'ADN simulées chacune pendant 100 nanosecondes de dynamique moléculaire après minimisation en utilisant a) des configurations entièrement élastiques et b) coudées, pour comparaison avec c) Carte de densité Cryo-EM à partir de reconstructions de phages asymétriques de P22 avec la densité de capside graphiquement supprimée. Crédit :Christopher G. Myers, B. Montgomery Pettitt, Branche médicale de l'Université du Texas
Le repliement du domaine de signalisation du facteur de transcription nucléaire n'est pas provoqué par des liaisons hydrogène et des effets hydrophobes, comme la plupart des molécules de protéines, selon Pettitt. Plutôt, quand les molécules plus longues trouvent trop de glycines dans un espace, ils dépassent leur solubilité et commencent à s'associer de manière inhabituelle.
"C'est comme mettre trop de sucre dans ton thé, " explique Pettitt. " Ça ne sera pas plus sucré. Le sucre doit tomber de la solution et trouver un partenaire - se précipitant en un morceau."
Écrire dans Science des protéines en 2015, il a décrit des simulations moléculaires réalisées sur Stampede qui ont aidé à expliquer comment et pourquoi les IDP s'effondrent en structures de type globule.
Les simulations ont calculé les forces des interactions dipôle-dipôle carbonyle (CO) - attractions entre l'extrémité positive d'une molécule polaire et l'extrémité négative d'une autre molécule polaire. Il a déterminé que ces interactions sont plus importantes dans l'effondrement et l'agrégation de longs brins de glycine que dans la formation de liaisons H.
"Étant donné que la colonne vertébrale est une caractéristique de toutes les protéines, Les interactions CO peuvent également jouer un rôle dans les protéines de séquence non triviale où la structure est finalement déterminée par l'emballage intérieur et les effets stabilisants des liaisons H et des interactions CO-CO, " a-t-il conclu.
La recherche a été rendue possible par une allocation de temps de calcul sur Stampede via l'Extreme Science and Engineering Discovery Environment (XSEDE) qui est soutenu par la National Science Foundation.
Pettit, champion de longue date du calcul intensif, n'utilise pas seulement les ressources TACC lui-même. Il encourage d'autres savants, y compris ses collègues du Sealy Center for Structural Biology and Molecular Biophysics, d'utiliser aussi des supercalculateurs.
« L'informatique avancée est importante pour l'analyse des données et le raffinement des données à partir d'expériences, Microscopie à rayons X et électronique, et informatique, " dit-il. " Tous ces problèmes ont des problèmes de traitement de données volumineux qui peuvent être résolus à l'aide de l'informatique de pointe. "
Lorsqu'il s'agit de percer les mystères de la biologie aux plus petites échelles, rien ne vaut un supercalculateur géant.