Des scientifiques de l'école de médecine Icahn du mont Sinaï, Sema4, et les institutions collaboratrices L'Université de New York et l'Université de Floride ont publié aujourd'hui un rapport détaillant leur nouveau méthode plus précise pour identifier les espèces et les souches microbiennes individuelles dans une communauté. Cette technique a des implications importantes pour l'analyse du microbiome, avec des applications potentielles à long terme pour les soins cliniques. Le journal est sorti aujourd'hui à Biotechnologie naturelle .
Les microbiomes sont des communautés de bactéries, virus, et d'autres microbes qui peuvent être trouvés partout, des surfaces des claviers et des téléphones portables aux environnements sur et en nous, comme notre bouche ou nos intestins. La perturbation du microbiome naturel a été impliquée dans des problèmes de santé, notamment les maladies infectieuses, cancéreux, et des troubles complexes tels que la maladie de Crohn, rectocolite hémorragique, et le diabète, parmi beaucoup d'autres. Une analyse réussie des microbiomes dépend de la capacité de zoomer sur ces communautés et d'identifier les espèces et les souches individuelles qui y vivent.
À ce jour, la plupart des techniques d'identification des membres microbiens de ces groupes fournissent une résolution insuffisante. Par exemple, une espèce ne peut être classée que dans le cadre de sa famille génétique plus large, plutôt que de s'identifier de manière unique. Les méthodes existantes ne sont pas non plus efficaces dans la caractérisation d'une classe importante de matériel génétique qui peut faire la navette entre différentes espèces bactériennes, appelés éléments génétiques mobiles.
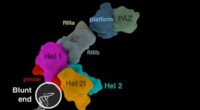
Dans ce nouveau travail, les scientifiques ont utilisé une molécule unique, Technologie de séquençage en temps réel et nouveaux outils de calcul pour classer les microbes pour la première fois en analysant à la fois leur code génétique et leurs schémas de méthylation, un deuxième code ADN qui régule l'activité des gènes. Cette approche plus globale utilisant le séquençage à lecture longue s'est avérée plus précise que les protocoles standard de l'industrie tels que le séquençage 16S ou le séquençage à lecture courte, corriger les erreurs et les résultats incomplets dans l'identification des microbes générés par ces méthodes. Surtout, la méthode fournit une nouvelle façon de lier des éléments génétiques mobiles à leurs hôtes bactériens, permettant aux scientifiques de prédire plus précisément la virulence, résistance aux antibiotiques, et d'autres traits biologiquement et cliniquement critiques d'espèces et de souches bactériennes individuelles.
« La communauté biomédicale a depuis longtemps besoin d'une méthode d'analyse du microbiome capable de résoudre des espèces et des souches individuelles avec une haute résolution, " dit Gang Fang, Doctorat, Professeur assistant de génétique et de sciences génomiques au Mont Sinaï, et auteur principal de l'article. "Nous avons découvert que les modèles de méthylation de l'ADN peuvent être exploités en tant que codes-barres naturels hautement informatifs pour aider à discriminer les espèces microbiennes les unes des autres, aider à associer des éléments génétiques mobiles à leurs génomes hôtes et à réaliser une analyse plus précise du microbiome. »
Dans des projets pilotes utilisant à la fois des échantillons de microbiome synthétiques et réels, les scientifiques ont pu faire la distinction entre des espèces et des souches de bactéries même étroitement apparentées. Ils ont utilisé des modèles de méthylation pour relier les données de séquences d'ADN connexes, fournissant des informations plus holistiques sur les organismes individuels. L'équipe a validé la méthode dans des communautés microbiennes de complexité faible à moyenne, et développe actuellement des technologies plus avancées pour résoudre efficacement les communautés très complexes telles que les microbiomes environnementaux.
"Ce projet démontre la sophistication et la puissance d'analyser de nombreux types de données ensemble pour fournir des informations qui ne sont pas possibles avec des approches plus simplistes, " a déclaré Eric Schadt, Doctorat, PDG de Sema4, Doyen de la médecine de précision au Mont Sinaï, et co-auteur de l'article. « La biologie est complexe, et nos analyses doivent représenter avec précision cette complexité si nous espérons éventuellement déployer ces informations à des fins cliniques. »