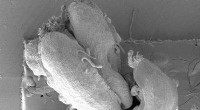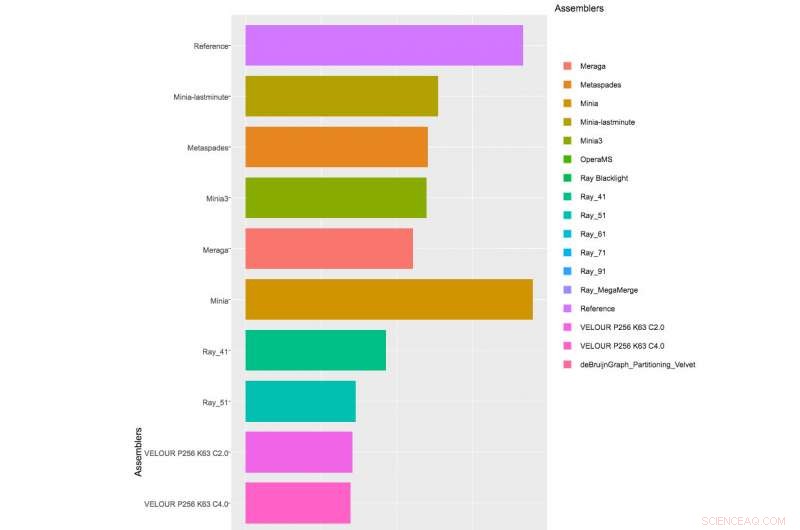
Tableau des résultats partiels des assembleurs appliqués au 1er Challenge CAMI, Ensemble de données 1. Cliquez ici pour voir le tableau complet.
Ils sont partout, mais invisible à l'oeil nu. Les microbes sont l'invisible, les forces influentes derrière la régulation des processus environnementaux clés tels que le cycle du carbone, pourtant la plupart d'entre eux restent inconnus. Depuis plus d'une décennie, le Département américain de l'énergie Joint Genome Institute (DOE JGI), une installation utilisateur du DOE Office of Science, a permis aux chercheurs d'étudier des microbes non cultivés incapables de se développer en laboratoire, en utilisant des approches de pointe telles que le séquençage génomique à haut débit des communautés environnementales ("métagénomique") et le développement d'outils informatiques pour découvrir et caractériser les communautés microbiennes de l'environnement. Pour s'attaquer à l'assemblage de métagénomes en un ensemble de segments d'ADN qui se chevauchent qui représentent ensemble une région consensus d'ADN ou de contigs, puis regrouper ces contigs dans des bacs génomiques, et enfin réaliser le profilage taxonomique des bacs génomiques, les analystes du monde entier ont développé une gamme d'outils de calcul différents, cependant, jusqu'à présent, il n'y avait pas de consensus sur la manière d'évaluer leur performance.
Publié le 2 octobre 2017 en Méthodes naturelles , une équipe comprenant des chercheurs du DOE JGI a décrit les résultats du défi d'évaluation critique de l'interprétation du métagénome (CAMI), le tout premier, évaluation comparative organisée par la communauté des outils de calcul pour les métagénomes. Le CAMI Challenge était dirigé par Alexander Sczyrba, chef du groupe de métagénomique computationnelle à l'Université de Bielefeld et ancien boursier postdoctoral du DOE JGI, et Alice McHardy, chef du laboratoire de recherche en biologie computationnelle des infections au Helmholtz Center for Infection Research.
« Il est très difficile pour les chercheurs de savoir quel programme utiliser pour un ensemble de données particulier et une analyse basée sur les résultats des documents de méthode, " a déclaré McHardy. " Les ensembles de données et les mesures d'évaluation utilisées dans les évaluations varient considérablement. Un autre problème est que les développeurs passent généralement beaucoup de temps à évaluer l'état de l'art lors de l'évaluation des performances d'un nouveau logiciel de cette manière. CAMI veut changer ces choses et implique la communauté dans la définition des normes et des meilleures pratiques pour l'évaluation et l'application de ces principes dans les défis d'analyse comparative."
Le Challenge CAMI s'est déroulé sur trois mois en 2015. Pour évaluer les outils de calcul, les organisateurs ont développé 3 ensembles de données de métagénome simulé en utilisant plus de 300 ébauches de génomes d'isolats bactériens et archéens séquencés et assemblés par le DOE JGI, qui faisaient partie du projet Genomic Encyclopedia of Bacteria and Archaeal publié récemment dans Biotechnologie naturelle . Ces génomes ont été partagés avec le consortium CAMI Challenges avant d'être rendus publics pour faciliter l'analyse comparative objective des différents outils. Les ensembles de données comprenaient également environ le même nombre de génomes de l'Institut Max Planck de Cologne, Allemagne, ainsi que des éléments circulaires et des virus. Les ensembles de données simulés étaient un ensemble de données échantillon unique de 15 milliards de bases (Go), un jeu de données de 40 Go avec 40 génomes et 20 éléments circulaires, et un ensemble de données de séries chronologiques de 75 Go composé de cinq échantillons et comprenant des centaines de génomes et d'éléments circulaires.
"JGI s'intéresse de près à l'analyse comparative d'outils et de technologies qui feraient progresser l'analyse des métagénomes et amélioreraient la qualité des données que nous fournissons aux utilisateurs. Après avoir publié la toute première étude sur l'utilisation d'ensembles de données simulés pour l'analyse comparative des outils de métagénomique de la JGI, c'est formidable de voir comment cette méthodologie s'est développée au fil des ans et maintenant grâce à cette étude, devenir un modèle d'efforts communautaires standardisés sur le terrain, " a déclaré Nikos Kyrpides, Chef de programme Super DOE JGI Prokaryote.
"JGI est très investi non seulement dans l'analyse comparative des protocoles de laboratoire, mais aussi des workflows de calcul, " a ajouté Tanja Woyke, responsable de la génomique microbienne du DOE JGI. " Cela rend notre participation aux efforts communautaires critiques tels que CAMI si importante. "
Avec plus de 40 équipes inscrites au Challenge, et les organisateurs du CAMI ont reçu 215 candidatures de 25 programmes à travers le monde, bien que seulement 17 équipes étaient disposées à publier leurs implémentations logicielles. Les organisateurs du CAMI ont évalué les outils de calcul dans 3 catégories. Une demi-douzaine d'assembleurs et de pipelines d'assemblage ont été évalués sur l'assemblage de séquences génomiques générées à partir de technologies de séquençage à lecture courte. Dans le défi du binning, five genome binners and 4 taxonomic binners were evaluated on criteria including the tools' efficacy in recovering individual genomes. Finalement, 10 taxonomic profilers with various parameter settings were evaluated on how well they could predict the identities and relative abundances of the microbes and circular elements. The benchmarking results are available on https://data.cami-challenge.org/results.
The CAMI organizers are already planning future benchmarking challenges, for example to evaluate and aid method development for long read sequencing technologies. "CAMI is an ongoing initiative, " noted Sczyrba. "We are currently further automating the benchmarking and comparative result visualizations. And we invite everyone interested to join and work with CAMI on providing comprehensive performance overviews of the computational metagenomics toolkit, to inform developers about current challenges in computational metagenomics and applied scientists of the most suitable software for their research questions."