Utiliser l'apprentissage automatique pour assister la conception moléculaire. Crédit :Wenbo Sun, Avancées scientifiques, doi:10.1126/sciadv.aay4275
Synthétiser des matériaux haute performance pour le photovoltaïque organique (OPV) qui convertissent le rayonnement solaire en courant continu, les scientifiques des matériaux doivent établir de manière significative la relation entre les structures chimiques et leurs propriétés photovoltaïques. Dans une nouvelle étude sur Avancées scientifiques , Wenbo Sun et une équipe comprenant des chercheurs de la School of Energy and Power Engineering, École d'automatisation, L'informatique, Génie électrique et technologies vertes et intelligentes, créé une nouvelle base de données de plus de 1, 700 documents de donateurs utilisant des rapports de littérature existants. Ils ont utilisé l'apprentissage supervisé avec des modèles d'apprentissage automatique pour établir des relations structure-propriété et des matériaux OPV à écran rapide en utilisant une variété d'entrées pour différents algorithmes de ML.
En utilisant des empreintes moléculaires (codant une structure d'une molécule en bits binaires) au-delà d'une longueur de 1000 bits Sun et al. obtenu une haute précision de prédiction ML. Ils ont vérifié la fiabilité de l'approche en examinant 10 matériaux donneurs nouvellement conçus pour la cohérence entre les prédictions du modèle et les résultats expérimentaux. Les résultats de ML ont présenté un outil puissant pour présélectionner de nouveaux matériaux OPV et accélérer le développement des OPV en ingénierie des matériaux.
Les cellules photovoltaïques organiques (OPV) peuvent faciliter la transformation directe et rentable de l'énergie solaire en électricité avec une croissance récente rapide pour dépasser les taux d'efficacité de conversion de puissance (PCE). La recherche traditionnelle sur le VPO s'est concentrée sur l'établissement d'une relation entre les nouvelles structures moléculaires du VPO et leurs propriétés photovoltaïques. Le processus traditionnel implique généralement la conception et la synthèse de matériaux photovoltaïques pour l'assemblage/l'optimisation de cellules photovoltaïques. De telles approches entraînent des cycles de recherche longs qui nécessitent un contrôle délicat de la synthèse chimique et de la fabrication des dispositifs, étapes expérimentales et purification. Le processus de développement actuel du VPO est lent et inefficace avec moins de 2000 molécules donneuses de VPO synthétisées et testées à ce jour. Cependant, les données recueillies à partir de décennies de travaux de recherche sont inestimables, avec des valeurs potentielles restant à explorer pleinement pour générer des matériaux OPV haute performance.
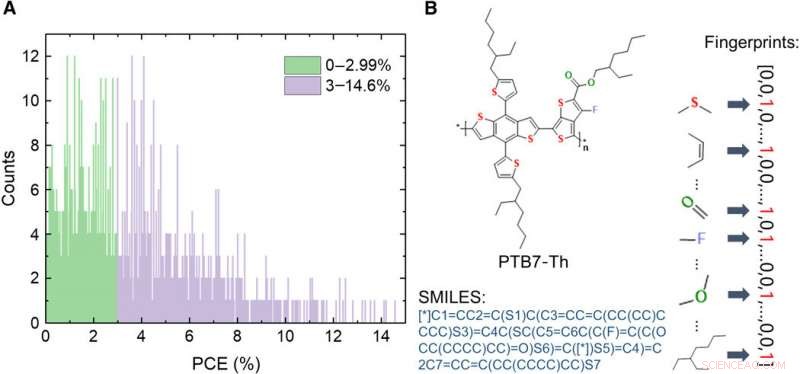
Informations sur la base de données des matériaux des donneurs de VPO. (A) Distribution des valeurs PCE des 1719 molécules dans la base de données. (B) Schémas d'expressions d'une molécule, y compris l'image, système simplifié d'entrée de ligne à entrée moléculaire (SMILES), et les empreintes digitales. Crédit :Avancées scientifiques, doi:10.1126/sciadv.aay4275
Pour extraire des informations utiles des données, Soleil et al. nécessitait un programme sophistiqué pour parcourir un vaste ensemble de données et extraire des relations parmi les entités. Étant donné que l'apprentissage automatique (ML) fournit des outils de calcul pour apprendre et reconnaître des modèles et des relations à l'aide d'un ensemble de données d'entraînement, l'équipe a utilisé une approche basée sur les données pour permettre l'apprentissage automatique et prédire diverses propriétés des matériaux. L'algorithme ML n'a pas eu à comprendre la chimie ou la physique derrière les propriétés des matériaux pour accomplir les tâches. Des méthodes similaires ont récemment prédit l'activité/les propriétés des matériaux avec succès lors de la découverte de matériaux, développement de médicaments et conception de matériaux. Avant les applications ML, les scientifiques avaient généré la chimioinformatique pour établir une boîte à outils utile.
Les scientifiques des matériaux n'ont exploré que récemment les applications de la ML dans le domaine de l'OPV. Dans le travail present, Soleil et al. a établi une base de données contenant 1719 matériaux de VPO de donneurs testés expérimentalement et recueillis dans la littérature. Ils ont d'abord étudié l'importance de l'expression du langage de programmation des molécules pour comprendre les performances du ML. Ils ont ensuite testé plusieurs types d'expressions, notamment des images, chaînes ASCII, deux types de descripteurs et sept types d'empreintes moléculaires. Ils ont observé que les prédictions du modèle étaient en bon accord avec les résultats expérimentaux. Les scientifiques s'attendent à ce que la nouvelle approche accélère considérablement le développement de nouveaux matériaux semi-conducteurs organiques hautement efficaces pour les applications de recherche OPV.
L'équipe de recherche a d'abord transformé les données brutes en une représentation lisible par machine. Il existe une variété d'expressions pour la même molécule comprenant des informations chimiques très différentes présentées à différents niveaux abstraits. En utilisant un ensemble de modèles ML, Soleil et al. ont exploré diverses expressions d'une molécule en comparant leur précision prédite pour l'efficacité de conversion de puissance (PCE) afin d'obtenir une précision de modèle d'apprentissage en profondeur de 69,41 %. La performance relativement insatisfaisante était due à la petite taille de la base de données. Par exemple, auparavant lorsqu'un même groupe utilisait un plus grand nombre de molécules allant jusqu'à 50, 000, la précision du modèle d'apprentissage en profondeur dépassait 90 pour cent. Pour former complètement un modèle d'apprentissage en profondeur, les chercheurs doivent mettre en place une base de données plus importante contenant des millions d'échantillons.
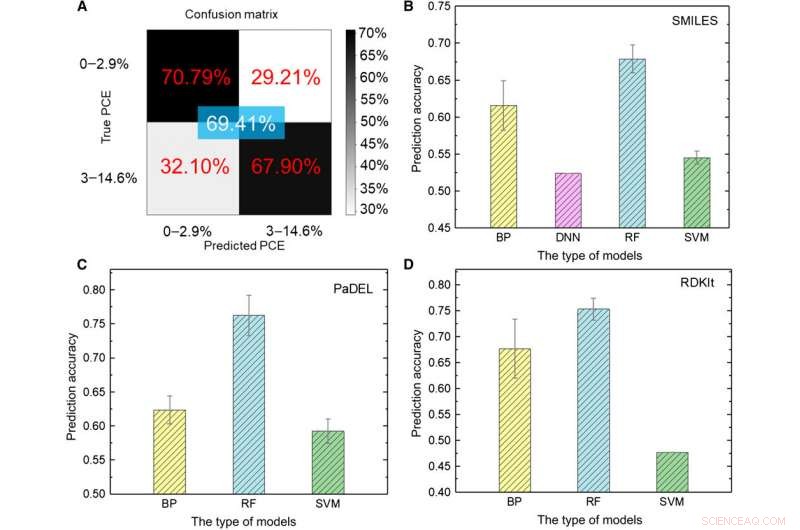
Résultats des tests de modèles ML. (A) Test du modèle d'apprentissage en profondeur en utilisant des images en entrée. (B à D) Tester les résultats de différents modèles de ML à l'aide de (B) SMILES, (C) PADEL, et (D) des descripteurs RDKIt en entrée. Crédit :Avancées scientifiques, doi:10.1126/sciadv.aay4275
Soleil et al. n'avait que des centaines de molécules dans chaque catégorie à l'heure actuelle, ce qui rend difficile pour le modèle d'extraire suffisamment d'informations pour une plus grande précision. Bien qu'il soit possible d'affiner un modèle pré-entraîné pour réduire la quantité de données requises, des milliers d'échantillons sont encore nécessaires pour accomplir un nombre suffisant de fonctionnalités. Cela a conduit à la possibilité d'augmenter la taille de la base de données lors de l'utilisation d'images pour exprimer des molécules.
Les scientifiques ont utilisé cinq types d'algorithmes de ML supervisés dans l'étude, y compris (1) réseau de neurones à propagation arrière (BP) (BPNN), (2) réseau de neurones profonds (DNN), (3) apprentissage en profondeur, (4) machine à vecteurs de support (SVM) et (5) forêt aléatoire (RF). Il s'agissait d'algorithmes avancés, où BPNN, Le DNN et l'apprentissage en profondeur étaient basés sur le réseau neutre artificiel (ANN). Le code SMILES (Simplified Molecular-input Line Entry System) a fourni une autre expression originale d'une molécule, dont Sun et al. utilisé comme entrées pour quatre modèles. Sur la base des résultats, la précision la plus élevée avoisinait 67,84 % pour le modèle RF. Comme avant, contrairement à l'apprentissage en profondeur, les quatre méthodes classiques ne pouvaient pas extraire les caractéristiques cachées. Dans son ensemble, Les SMILES ont été moins performants que les images en tant que descripteurs de molécules pour prédire la classe PCE (efficacité de conversion de puissance) dans les données.
Les chercheurs ont ensuite utilisé des descripteurs moléculaires qui peuvent décrire les propriétés d'une molécule à l'aide d'un ensemble de nombres au lieu de l'expression directe d'une structure chimique. L'équipe de recherche a utilisé deux types de descripteurs PaDEL et RDKIt dans l'étude. Après des analyses approfondies sur tous les modèles de ML, une grande taille de données impliquait plus de descripteurs non pertinents pour PCE affectant les performances ANN. Relativement, une petite taille de données impliquait des informations chimiques inefficaces pour entraîner efficacement les modèles ML, lors de l'utilisation de descripteurs moléculaires comme entrée dans les approches ML, la clé reposait sur la recherche de descripteurs appropriés directement liés à l'objet cible.
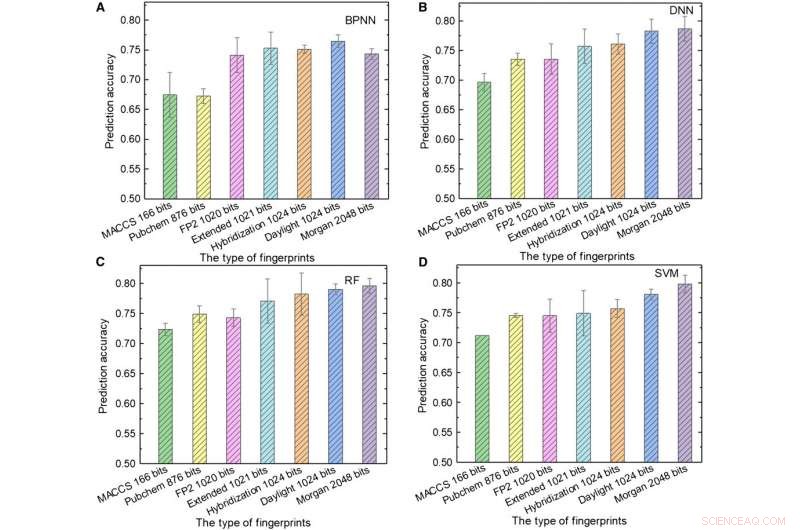
Performances des modèles ML. (A à D) Les résultats des tests de (A) BPNN, (B) DNN, (C) RF, et (D) SVM utilisant différents types d'empreintes digitales en entrée. Crédit :Avancées scientifiques, doi:10.1126/sciadv.aay4275.
L'équipe a ensuite utilisé des empreintes digitales moléculaires; généralement conçu pour représenter les molécules en tant qu'objets mathématiques et créé à l'origine pour identifier les isomères. Lors du filtrage de la base de données à grande échelle, le concept est représenté par un tableau de bits contenant des "1" et des "0" pour décrire la présence ou l'absence de sous-structures ou de motifs spécifiques au sein des molécules. Soleil et al. ont utilisé sept types d'empreintes digitales comme entrées pour entraîner les modèles ML et ont examiné l'influence de la longueur des empreintes digitales sur les performances de prédiction de différents modèles pour obtenir diverses empreintes digitales. Par exemple, les empreintes digitales du système d'accès moléculaire (MACCS) contenaient 166 bits et constituaient l'entrée la plus courte et les résultats n'étaient pas satisfaisants en raison de leurs informations limitées.
Soleil et al. a montré la meilleure combinaison de langage de programmation et d'algorithme ML obtenue en utilisant des empreintes d'hybridation de 1024 bits et RF, atteindre une précision de prédiction de 81,76 % ; où les empreintes d'hybridation représentaient les états d'hybridation SP2 des molécules. Lorsque la longueur de l'empreinte digitale est passée de 166 à 1024 bits, les performances de tous les modèles ML se sont améliorées puisque les empreintes digitales plus longues incluaient plus d'informations chimiques.
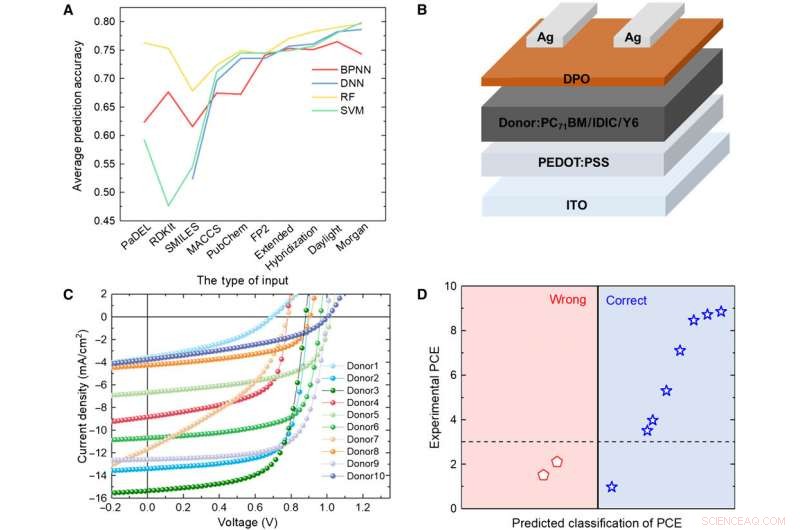
Vérification des modèles ML avec expérimentation. (A) Comparaison des résultats de quatre modèles différents. (B) Schéma de principe de l'architecture cellulaire utilisée dans cette étude. (C) Courbe J-V de la cellule solaire avec la couche active en utilisant le matériau donneur prévu. (D) Résultats de prédiction par rapport aux données expérimentales pour les matériaux donneurs prédits avec l'algorithme RF et les empreintes digitales Daylight. Crédit :Avancées scientifiques, doi:10.1126/sciadv.aay4275.
Pour tester la fiabilité des modèles ML, Soleil et al. synthétisé 10 nouvelles molécules donneuses de VPO. Ensuite, nous avons utilisé trois empreintes digitales représentatives pour exprimer la structure chimique des nouvelles molécules et comparé les résultats prédits par le modèle RF et les valeurs expérimentales du PCE. Le système a classé huit des 10 molécules. Les résultats ont indiqué le potentiel des matériaux synthétiques pour les applications OPV avec une optimisation expérimentale supplémentaire pour deux des nouveaux matériaux. Un changement mineur de structure pourrait entraîner une grande différence dans les valeurs de PCE. De façon encourageante, les modèles ML ont identifié ces modifications mineures pour faciliter des résultats de prédiction favorables.
De cette façon, Wenbo Sun et ses collègues ont utilisé une base de données documentaire sur les matériaux des donneurs de VPO et une variété d'expressions de langage de programmation (images, chaînes ASCII, descripteurs et empreintes moléculaires) pour construire des modèles ML et prédire la classe OPV PCE correspondante. L'équipe a démontré un schéma pour concevoir des matériaux donneurs de VPO à l'aide d'approches ML et d'analyses expérimentales. Ils ont présélectionné un grand nombre de matériaux donneurs en utilisant le modèle ML pour identifier les principaux candidats pour la synthèse et d'autres expériences. Le nouveau travail peut accélérer la conception de nouveaux matériaux donneurs pour accélérer le développement de VPO à PCE élevé. L'utilisation de ML en conjonction avec des expériences fera progresser la découverte de matériaux.
© 2019 Réseau Science X