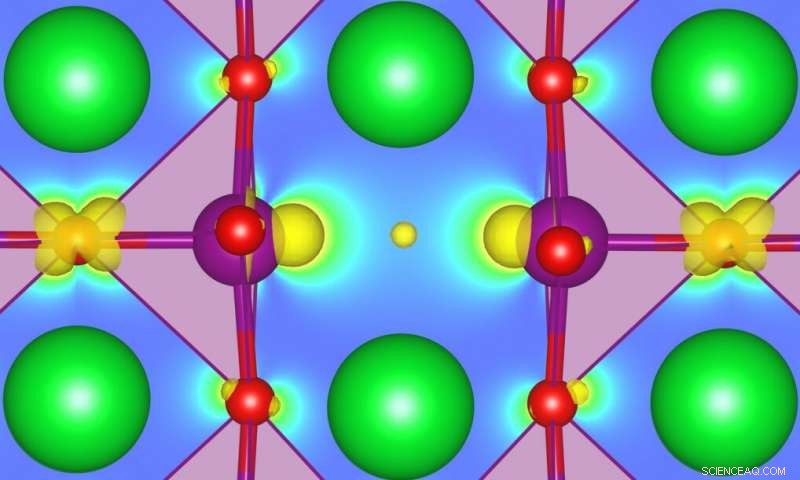
Comprendre comment les défauts peuvent affecter les propriétés de l'état fondamental, favoriser les transitions de phase, ou permettre des fonctionnalités entièrement nouvelles dans certains oxydes fortement corrélés est devenu un sujet d'intérêt majeur dans le domaine de la conception et de la découverte de nouveaux matériaux fonctionnels. SrMnO3 (SMO) est un exemple particulièrement intéressant, mais une meilleure caractérisation est nécessaire. Les chercheurs de MARVEL ont maintenant développé une méthode qui peut conduire à des prédictions plus précises de l'énergie des défauts associés aux états in-gap dans les semi-conducteurs ou les isolants. Crédit :Ulrich Aschauer
Comprendre comment les défauts peuvent affecter les propriétés de l'état fondamental, favoriser les transitions de phase, ou permettre des fonctionnalités entièrement nouvelles dans certains oxydes fortement corrélés est devenu un sujet d'intérêt majeur dans le domaine de la conception et de la découverte de nouveaux matériaux fonctionnels. SrMnO
Certains oxydes de pérovskite, par exemple, ont montré un large éventail de propriétés fonctionnelles pertinentes sur le plan technologique, telles que la ferroélectricité et le magnétisme, qui peuvent être réglées par contrainte. Souche, cependant, se couple également avec la chimie des défauts pour déterminer les propriétés du matériau.
SrMnO
Ces prédictions précédentes étaient cependant basées sur des calculs de la théorie de la fonctionnelle de la densité (DFT) qui incorporaient une correction U basée sur les propriétés électroniques et magnétiques des manganites stoechiométriques. Alors que l'inclusion de U, destinée à corriger l'auto-interaction des électrons dans les oxydes complexes, est nécessaire dans de tels matériaux, le choix spécifique de U basé sur les propriétés stoechiométriques du matériau pourrait conduire à des lacunes potentielles dans la description des SMO défectueux - les ions manganèse autour du défaut ont un environnement de coordination différent.
Selon l'état de charge du défaut, un problème supplémentaire est lié à la description des multiples états d'oxydation présents dans le SMO défectueux. La formation de lacunes d'oxygène est généralement compensée en charge par une réduction de l'état d'oxydation (OS) des ions manganèse adjacents à la lacune, qui peut donc ne pas être correctement décrit par le même U.
C'est pourquoi la postdoctorante de l'Université de Berne Chiara Ricca et ses collègues ont décidé qu'il était essentiel de prendre en compte les effets structuraux et chimiques locaux pour chaque site de métal de transition dans l'oxyde lors de la recherche d'une description précise du SMO défectueux. En collaboration avec une équipe du laboratoire THEOS de Nicola Marzari, qui a récemment développé une approche basée sur la théorie des perturbations fonctionnelles de la densité (DFPT) pour calculer les paramètres U, ils ont utilisé des valeurs U dépendant du site auto-cohérentes calculées à partir des premiers principes pour étudier la chimie des défauts et les propriétés magnétiques des films minces SMO en vrac et contraints.
« Cette collaboration extrêmement étroite entre les deux groupes, l'une axée sur le développement de méthodes et l'autre sur les applications dans les matériaux oxydes défectueux, a été suscitée par l'union de ces différents pôles de recherche sous l'égide de MARVEL », a déclaré Ulrich Aschauer de l'Université de Berne, l'un des deux IP impliqués dans les travaux.
Les résultats montrent que ce U auto-cohérent améliore la structure du SrMnO stoechiométrique
"Nous pensons que cette approche peut conduire à des prédictions plus précises de l'énergétique des défauts associés aux états in-gap dans les semi-conducteurs ou les isolants à la fois par rapport à la DFT standard et éventuellement aux fonctionnelles hybrides à un coût de calcul nettement inférieur à celui de ces dernières, " a déclaré Ricca. "C'est grâce à une description appropriée des effets chimiques structuraux et locaux induits par les défauts."