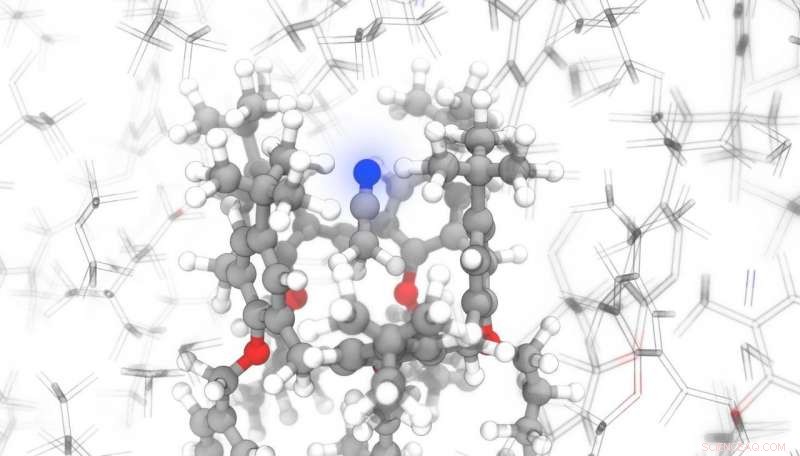
Crédit :Michèle Ceriotti / EPFL
De nos jours, de nombreux médicaments sont produits sous forme de solides en poudre. Mais pour bien comprendre comment les principes actifs vont se comporter une fois à l'intérieur du corps, les scientifiques doivent connaître leur structure exacte au niveau atomique. Par exemple, la façon dont les molécules sont disposées à l'intérieur d'un cristal a un impact direct sur les propriétés d'un composé, comme sa solubilité. Les chercheurs travaillent donc dur pour développer des technologies capables d'identifier facilement les structures cristallines exactes des poudres microcristallines.
Une équipe de scientifiques de l'EPFL a maintenant écrit un programme d'apprentissage automatique qui peut prédire, en un temps record, comment les atomes réagiront à un champ magnétique appliqué. Cela peut être combiné avec la spectroscopie par résonance magnétique nucléaire (RMN) pour déterminer l'emplacement exact des atomes dans les composés organiques complexes. Cela peut être très avantageux pour les entreprises pharmaceutiques, qui doivent surveiller attentivement les structures de leurs molécules pour répondre aux exigences de sécurité des patients. Leurs recherches ont été publiées dans Communication Nature .
Des vitesses vertigineuses avec l'IA
La spectroscopie RMN est une méthode bien connue et très efficace pour sonder les champs magnétiques entre les atomes et déterminer comment les atomes voisins interagissent les uns avec les autres. Cependant, la détermination complète de la structure cristalline par spectroscopie RMN nécessite des opérations extrêmement compliquées, calculs chronophages impliquant la chimie quantique - presque impossible pour des molécules avec des structures très complexes.
Mais le programme développé à l'EPFL permet de surmonter ces obstacles. Les scientifiques ont formé leur modèle d'IA sur des structures moléculaires extraites de bases de données structurelles. "Même pour des molécules relativement simples, ce modèle est presque 10, 000 fois plus rapide que les méthodes existantes, et l'avantage grandit énormément lorsqu'on considère des composés plus complexes, " dit Michèle Ceriotti, responsable du Laboratoire de science computationnelle et modélisation de l'École d'ingénieurs de l'EPFL et co-auteur de l'étude. "Pour prédire la signature RMN d'un cristal avec près de 1, 600 atomes, notre technique – ShiftML – nécessite environ six minutes; le même exploit aurait pris 16 ans avec des techniques conventionnelles."
Ce nouveau programme permettra d'utiliser des approches complètement différentes qui seront plus rapides et permettront d'accéder à des molécules plus grosses. "C'est vraiment excitant car l'accélération massive des temps de calcul nous permettra de couvrir des espaces conformationnels beaucoup plus grands et de déterminer correctement des structures là où cela n'était tout simplement pas possible auparavant. Cela met la plupart des molécules médicamenteuses contemporaines complexes à portée de main, " dit Lyndon Emsley, responsable du Laboratoire de résonance magnétique de la Faculté des sciences fondamentales de l'EPFL et co-auteur de l'étude.
Le programme est maintenant disponible gratuitement en ligne. « Tout le monde peut télécharger une molécule et obtenir sa signature RMN en quelques minutes, " dit Ceriotti.