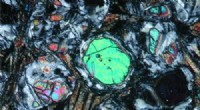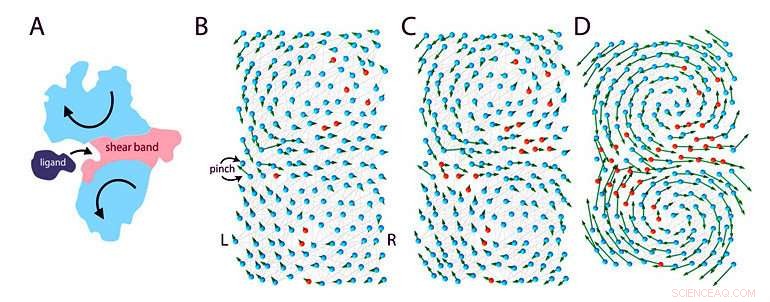
Figure 1 :Modèle élastique d'une protéine se liant à un ligand. (A) Lorsqu'une protéine se lie à un ligand, il subit un mouvement à grande échelle (flèches) qui sont les signatures des protéines fonctionnelles de flexion. Ceci n'est possible que grâce à la présence de certaines régions « floppy » (« bande de cisaillement » rose) à travers la protéine qui séparent les régions rigides (bleues) de la protéine en deux domaines. (B)-(D) L'équipe a modélisé une protéine de 200 acides aminés au cours de différentes étapes d'évolution :passage d'un état non fonctionnel (B) à un état fonctionnel (D). La protéine est modélisée comme un réseau de ressorts élastiques avec deux types d'acides aminés, modelé sous forme de billes :les acides aminés roses sont flexibles et les acides aminés bleus sont rigides. Les chercheurs imitent l'évolution en changeant un acide aminé aléatoire à la fois (mutation) du rose au bleu. Initialement, la protéine est pour la plupart rigide et non fonctionnelle. Au cours de l'évolution, des acides aminés flexibles sont ajoutés, certains utiles, d'autres non. Heures supplémentaires, une région « disquette » se forme au centre de la molécule, ce qui rend la protéine plus flexible pour se plier et se lier au ligand. Le modèle a estimé qu'une solution efficace est atteinte après un millier de mutations. Crédit : Institut des sciences fondamentales
Une équipe internationale a développé un modèle qui simule l'évolution des protéines. En partant de raide, protéines non fonctionnelles, le modèle informatique montre comment les composants protéiques en évolution peuvent travailler ensemble pour donner naissance à des machines moléculaires dynamiques et efficaces. La flexibilité permet aux protéines de changer leur conformation 3D pour se lier à d'autres molécules :cette propriété est cruciale pour leur fonction. Prof. Tsvi Tlusty et Dr Sandipan Dutta au Center for Soft and Living Matter, au sein de l'Institut des sciences fondamentales (IBS, Corée du Sud), en collaboration avec le professeur Albert Libchaber de l'Université Rockefeller et le professeur Jean-Pierre Eckmann de l'Université de Genève, ont mimé l'évolution des gènes pour obtenir des protéines capables de se plier et de se lier à d'autres molécules. La compréhension de cette relation est l'un des aspects les plus recherchés de la biologie des protéines; cela pourrait aider à expliquer l'action pharmaceutique des médicaments se liant à leurs cibles.
L'évolution a façonné le monde vivant que nous voyons autour de nous depuis des milliards d'années. Des millions de protéines fonctionnent harmonieusement pour maintenir ces processus vitaux. Ils sont responsables du bon fonctionnement de tout organisme :ils reconnaissent d'autres molécules (ligands), lier à eux et les convertir. D'autres ont une fonction de transport, structurer, et le soutien aux cellules. Les gènes stockent les informations sur la production et la conception de ces machines moléculaires. Cependant, malgré des décennies de recherche, dresser la « carte » qui trace le chemin des gènes à la fonction protéique n'est pas anodin.
Selon une hypothèse récente, la fonction des protéines repose sur des "articulations flexibles". Cette étude, Publié dans Actes de l'Académie nationale des sciences ( PNAS ), examine le lien entre fonction et flexibilité en modélisant des protéines telles que des réseaux élastiques. Dans ce modèle, les protéines sont constituées d'acides aminés flexibles (polaires) et rigides (hydrophobes) reliés par des "ressorts" moléculaires. Si certaines régions de la protéine sont suffisamment flexibles, ils forment un canal "disquette", et toute la machine moléculaire peut se plier comme une charnière. Ce mouvement leur permet de se lier efficacement à d'autres molécules. La liaison entre un ligand et une protéine rigide ou flexible peut être considérée comme une balle atterrissant sur un rocher ou un oreiller mou. La balle est susceptible de rebondir après avoir heurté le rocher, mais l'oreiller est plus susceptible de l'accepter. Par conséquent, la protéine flexible est un meilleur liant.
Dans ce modèle, les gènes stockent les détails de la conception de la protéine de manière binaire :les acides aminés flexibles sont stockés sous forme de zéros et les acides aminés rigides sous forme de uns. Par conséquent, la structure entière de la protéine peut être simplifiée sous forme de code, comme 11110001...111, semblable à la mémoire numérique d'un ordinateur. Cependant, tous les codes ne donnent pas naissance à des protéines fonctionnelles, par exemple un code avec seulement des uns :111111…1111, donnerait lieu à une protéine entièrement raide, Incapable de bouger, et non fonctionnel. Parmi tous les codes possibles, seuls certains produisent une protéine fonctionnelle avec une région "floppy" au centre qui peut accueillir le ligand.
Le modèle imite l'évolution en changeant un acide aminé aléatoire à la fois. Au cours de l'évolution, les zéros et les uns dans le gène sont inversés au hasard à travers un processus appelé mutation. La plupart des mutations n'apportent aucune différence, ou conduire à des protéines non fonctionnelles, mais certaines mutations rares peuvent donner naissance à une protéine plus efficace. Essentiellement, des protéines fonctionnelles et non fonctionnelles sont produites au cours de l'évolution, mais selon la théorie de Darwin de la "survie du plus fort", seules les protéines fonctionnelles sont conservées et les protéines non fonctionnelles finissent par disparaître.
A quoi ressemble un code « fonctionnel » ? La réponse n'est pas simple. En réalité, le nombre de codes d'une protéine fonctionnelle, même une simple protéine, est énorme, plus grand que la taille de l'univers. Cependant, en utilisant des techniques d'analyse de données, il est possible de rechercher des motifs cachés dans tous les codes fonctionnels pour rechercher certaines caractéristiques unificatrices. Par exemple, le canal "floppy" dans la protéine a des caractéristiques intéressantes et particulières, et une mutation à une extrémité du canal a des effets à longue portée qui peuvent fortement affecter le maintien des mutations d'autres acides aminés distants.
"À l'avenir, nous prévoyons d'explorer comment appliquer cette étude à de vraies protéines, comme les kinases, " a déclaré le chef de groupe Tsvi Tlusty, un correspondant dans l'étude. "De plus, l'étude ouvre des pistes pour étudier l'évolution d'autres fonctions protéiques, comme la reconnaissance moléculaire. En utilisant d'énormes bases de données, qui ont été développés grâce à des années de recherche, peut probablement découvrir des phénomènes sous-jacents à l'évolution des protéines."