Déverrouiller des informations biologiques à partir de données génomiques unicellulaires complexes est devenu plus facile et plus précis, grâce à l'outil innovant scLENS développé par le groupe de mathématiques biomédicales au sein du Centre IBS pour les sciences mathématiques et informatiques dirigé par l'enquêteur en chef Kim Jae Kyoung, qui est également professeur au KAIST. Cela représente un pas en avant significatif dans le domaine de la transcriptomique unicellulaire.
La recherche est publiée dans la revue Nature Communications .
L'analyse génomique unicellulaire est une technique avancée qui mesure l'expression des gènes au niveau de chaque cellule, révélant des changements et des interactions cellulaires qui ne sont pas observables avec les méthodes d'analyse génomique traditionnelles. Lorsqu'elle est appliquée aux tissus cancéreux, cette analyse peut délimiter la composition de divers types de cellules au sein d'une tumeur, fournissant ainsi un aperçu de la progression du cancer et identifiant les gènes clés impliqués à chaque étape de progression.
Malgré l’immense potentiel de l’analyse génomique unicellulaire, la gestion de la grande quantité de données qu’elle génère a toujours été un défi. La quantité de données couvre l’expression de dizaines de milliers de gènes dans des centaines, voire des milliers de cellules individuelles. Cela entraîne non seulement de grands ensembles de données, mais introduit également des distorsions liées au bruit, qui surviennent en partie à cause des limitations actuelles des mesures.
L'auteur correspondant Kim Jae Kyoung a souligné :« Il y a eu des progrès remarquables dans les technologies expérimentales pour analyser les transcriptomes unicellulaires au cours de la dernière décennie. Cependant, en raison des limites des méthodes d'analyse des données, il a été difficile d'utiliser pleinement les données précieuses obtenues grâce à ces méthodes. coût et temps considérables."
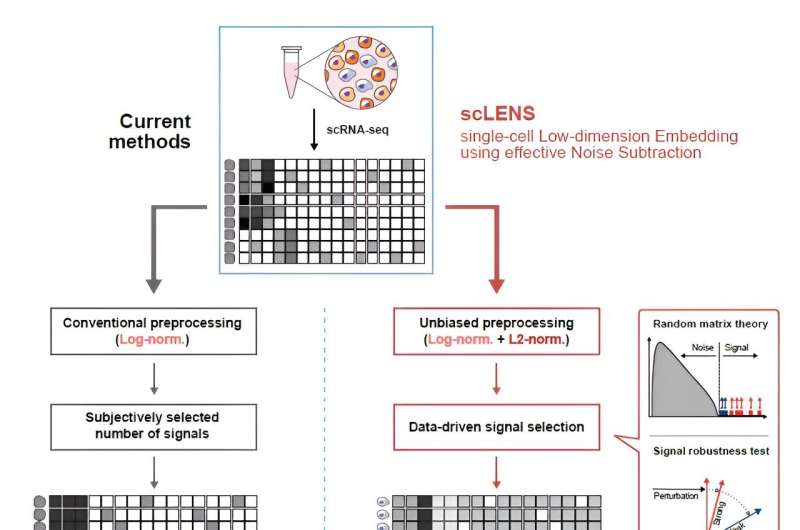
Les chercheurs ont développé au fil des années de nombreuses méthodes d’analyse pour discerner les signaux biologiques de ce bruit. Cependant, la précision de ces méthodes est loin d’être satisfaisante. Un problème crucial est que la détermination des seuils de signal et de bruit dépend souvent de décisions subjectives des utilisateurs.
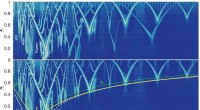
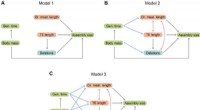
Le nouvel outil scLENS exploite la théorie des matrices aléatoires et le test de robustesse du signal pour différencier automatiquement les signaux du bruit sans s'appuyer sur les entrées subjectives de l'utilisateur.
Le premier auteur, Kim Hyun, a déclaré :« Auparavant, les utilisateurs devaient décider arbitrairement du seuil de signal et de bruit, ce qui compromettait la reproductibilité des résultats d'analyse et introduisait de la subjectivité. scLENS élimine ce problème en détectant automatiquement les signaux en utilisant uniquement la structure inhérente des données. »
Au cours du développement de scLENS, les chercheurs ont identifié les raisons fondamentales des inexactitudes dans les méthodes d’analyse existantes. Ils ont constaté que les méthodes de prétraitement des données couramment utilisées déforment à la fois les signaux biologiques et le bruit. La nouvelle approche de prétraitement proposée par scLENS est exempte de telles distorsions.
En résolvant les problèmes liés au seuil de bruit déterminé par le choix subjectif de l'utilisateur et à la distorsion du signal dans le prétraitement conventionnel des données, scLENS surpasse considérablement les méthodes existantes en termes de précision. De plus, scLENS automatise le processus laborieux de sélection des dimensions du signal, permettant aux chercheurs d'extraire les signaux biologiques de manière pratique et automatique.
Ci Kim a ajouté :« scLENS résout des problèmes majeurs dans l'analyse des données du transcriptome unicellulaire, en améliorant considérablement la précision et l'efficacité tout au long du processus d'analyse. Il s'agit d'un excellent exemple de la façon dont les théories mathématiques fondamentales peuvent stimuler l'innovation dans la recherche en sciences de la vie, permettant aux chercheurs d'approfondir Répondez rapidement et précisément aux questions biologiques et découvrez des secrets de la vie qui étaient auparavant cachés. "
Plus d'informations : Hyun Kim et al, scLENS :détection de signaux basée sur les données pour une analyse impartiale des données scRNA-seq, Nature Communications (2024). DOI :10.1038/s41467-024-47884-3
Informations sur le journal : Communications naturelles
Fourni par l'Institut des sciences fondamentales