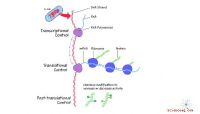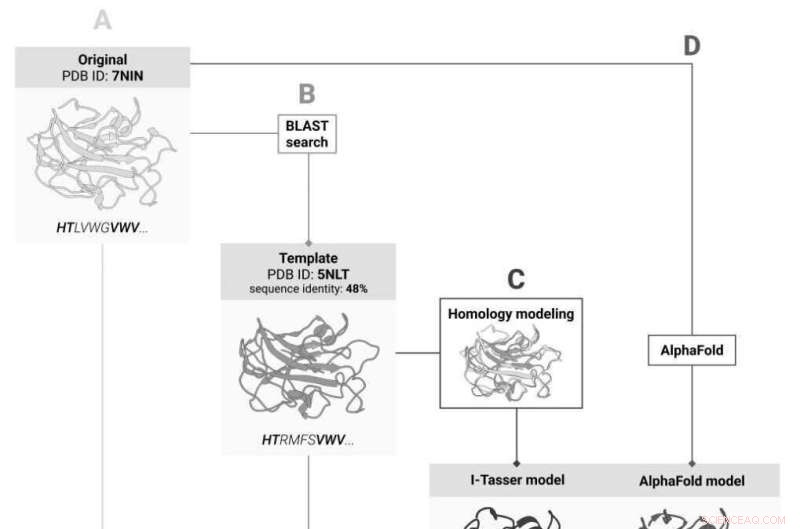
Quatre façons de prédire les changements dans la stabilité des protéines après mutation :(A) par la structure de la protéine d'origine; (B) par la structure de son homologue; (C) par la structure de la protéine d'origine prédite sur la base de la structure de l'holomlog, et (D) par la structure prédite par l'intelligence artificielle sur la base de la séquence d'acides aminés. Crédit :Institut des sciences et technologies de Skolkovo
Des chercheurs du Skoltech Center for Molecular and Cellular Biology ont comparé différentes méthodes de prédiction de la structure des protéines en termes d'évaluation de la stabilité des protéines mutantes et ont obtenu le même résultat pour les structures prédites par l'IA et les protéines expérimentales tridimensionnelles (3D) avec des séquences d'acides aminés similaires. Cependant, la tentative de prédire la structure de la protéine ciblée à partir de la structure connue de son "parent" n'a fait que rendre la prédiction moins précise. Les découvertes de l'équipe faciliteront les calculs préliminaires dans l'évaluation des changements de stabilité provoqués par la mutation. La recherche a été publiée dans Bioinformatics .
Les expériences biologiques impliquent souvent des protéines mutantes, nécessaires à l'étude de la structure et des fonctions des protéines ou des processus cellulaires, ainsi qu'à l'ingénierie des protéines. Les mutations sont connues pour affecter la structure et la stabilité d'une protéine. Étant donné que les expériences sont trop coûteuses et prennent trop de temps, les scientifiques créent une solution de contournement sous la forme de méthodes de calcul pour évaluer l'impact des mutations sur la stabilité. Cependant, leurs applications nécessitent la connaissance de la structure 3D d'une protéine.
Une structure 3D expérimentale n'est pas disponible pour toutes les protéines et est susceptible de manquer pour celle ciblée par l'équipe. Si tel est le cas, les modèles 3D des homologues de la protéine, c'est-à-dire ses "parents les plus proches", peuvent fournir la bouée de sauvetage, car le degré de similitude dans les séquences d'acides aminés qui assure une bonne correspondance entre les structures 3D des protéines est bien connu. La solution serait d'abord de prédire la structure de la protéine en fonction de la structure connue de son homologue, puis de calculer l'impact des mutations pour le modèle prédit.
Grâce à la percée de l'année dernière dans la prédiction de la structure des protéines, les scientifiques ont maintenant une alternative :au lieu de prédire la structure 3D basée sur des homologues, ils peuvent utiliser l'outil AlphaFold basé sur l'IA qui prédit la structure de la protéine à partir de la séquence d'acides aminés et a déjà traité avec la grande majorité des protéines connues à ce jour.
Dans leur étude récente, les chercheurs de Skoltech ont décidé de déterminer laquelle de ces approches fonctionne le mieux pour prédire les changements de stabilité lors d'une mutation. Aussi précis qu'AlphaFold puisse être, trouver la structure de la protéine par des expériences reste toujours le "gold standard". Lors de la comparaison des deux approches, l'équipe a utilisé sept méthodes d'évaluation de la stabilité et a comparé leurs résultats à ceux d'AlphaFold et d'I-Tasser, le meilleur système de prédiction de structure basé sur les homologues. En outre, les chercheurs ont vérifié s'ils pouvaient ignorer la prédiction de la structure basée sur les homologues et calculer la stabilité de la structure connue de la protéine homologue.
"Nous avons décidé de découvrir jusqu'où nous nous écarterions de la prédiction précise si nous utilisions la structure protéique "voisine" au lieu de la vraie. Il s'est avéré que l'étape de prédiction basée sur l'homologie ne fait qu'empirer les choses en donnant un résultat moins précis. Nous avons montré que cela ne fait pratiquement aucune différence que vous utilisiez la structure expérimentale de l'homologue ou la prédiction d'AlphaFold. En un sens, il s'agissait de validation :face à une nouvelle méthode, vous devez d'abord vérifier si elle fonctionne pour votre tâche. . C'est exactement ce que nous avons fait », a déclaré le premier auteur de l'étude, Skoltech Ph.D. étudiante Marina Pak, commentaires.
"Avec tout ce remue-ménage autour d'AlphaFold, certains scientifiques et non-professionnels pensent qu'il a résolu tous les problèmes de recherche sur les protéines en biologie computationnelle, mais ce n'est pas le cas. Par exemple, la prédiction des changements de stabilité induits par la mutation affiche toujours une fiabilité plutôt faible, même bien que le changement de stabilité soit parmi les principaux moteurs de la fonctionnalité des protéines. Un outil qui pourrait déterminer sans ambiguïté l'impact de la mutation sur la stabilité aiderait à la fois à planifier l'expérience et à interpréter les résultats. Supposons que pour une protéine qui n'est pas optimale en termes de stabilité, nous souhaitons trouver des mutations qui la rendraient stable dans les conditions souhaitées, par exemple, garantir qu'elle reste active à haute température. Une fois que nous pourrons le faire uniquement par des calculs, l'approche de la refonte et de l'optimisation des protéines changera radicalement », conclut l'auteur principal de l'étude, le professeur adjoint de Skoltech, Dmitry Ivankov.
Bien que la prédiction des changements de stabilité semble plus facile que la prédiction de la structure 3D, elle reste un défi insoluble, même pour l'IA. La rareté des données d'entraînement n'est qu'un des problèmes :AlphaFold avait près de 200 000 structures protéiques à entraîner, mais les données expérimentales sur les changements de stabilité s'élèvent à des milliers d'ensembles tout en ne couvrant que quelques dizaines de protéines uniques. Les auteurs espèrent que si davantage de données deviennent disponibles et que les chercheurs s'intéressent davantage à la tâche, une percée se produira certainement bientôt. Les physiciens utilisent l'IA pour trouver les nœuds protéiques les plus complexes à ce jour