Crédit :Meletios Verras/Shutterstock
Parfois, vous ne savez pas ce que vous cherchez jusqu'à ce que vous le trouviez; et cela est particulièrement vrai en ce qui concerne les énormes ensembles de données qui peuvent être générés à l'aide de techniques de séquençage modernes. Maintenant, des chercheurs du Japon rapportent le développement d'un cadre statistique qui peut effectuer une extraction impartiale des communications cellule-cellule biologiquement pertinentes à partir d'une mer de données spatiales sur l'expression des gènes.
Dans une étude publiée en septembre dans Bioinformatics , des chercheurs de l'Université de Tsukuba ont révélé qu'une nouvelle méthode d'analyse statistique peut identifier avec précision la communication cellule-cellule affectant l'expression des gènes au niveau de la cellule unique.
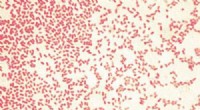
La communication entre les cellules régule l'expression des gènes de manière cruciale pour le fonctionnement normal ainsi que pour le développement de la maladie. Bien que le séquençage d'ARN unicellulaire et la transcriptomique résolue spatialement puissent fournir un aperçu de cette communication, les méthodes actuelles d'analyse de ces types de données présentent des limites importantes.
"La plupart des méthodes d'analyse statistique existantes ne tiennent pas compte de l'organisation spatiale des cellules au sein d'un organe composé de divers types de cellules", déclare le professeur agrégé Haruka Ozaki, auteur principal de l'étude. "Cependant, l'emplacement des cellules, le nombre de cellules et les types de cellules à proximité affectent l'expression des gènes dans les cellules voisines."
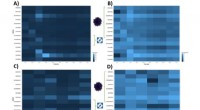
Pour capturer cette complexité, les chercheurs ont créé un cadre statistique appelé CCPLS (analyse des communications cellule-cellule par modélisation de régression partielle des moindres carrés) qui analyse les données spatiales d'expression génique à une résolution unicellulaire. L'objectif de ce système était d'identifier et de quantifier l'influence des types de cellules voisines sur la variabilité intercellulaire de l'expression génique.
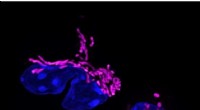
"Nous avons d'abord appliqué CCPLS à un ensemble de données simulées et avons constaté qu'il estimait avec précision les effets de plusieurs types de cellules voisines sur l'expression des gènes", explique le professeur agrégé Ozaki. "Ensuite, nous avons appliqué le système à un ensemble de données du monde réel et montré que les astrocytes favorisent la différenciation des cellules précurseurs d'oligodendrocytes en oligodendrocytes, ce qui est cohérent avec les expériences précédentes sur la souris."
Ensuite, CCPLS a été appliqué à un autre ensemble de données du monde réel contenant des données d'expression génique de neuf types de cellules différents trouvés dans le côlon. L'analyse a montré que le développement des cellules épithéliales des cellules B immatures se produit par la communication avec les cellules IgA B, ce qui n'a pas été signalé auparavant.
"Nos résultats montrent que le CCPLS peut être utilisé pour extraire des informations biologiquement pertinentes sur les communications cellule-cellule à partir d'ensembles de données complexes", déclare le professeur Ozaki.
Étant donné que CCPLS a surpassé un cadre statistique existant dans l'identification de la variabilité de l'expression génique régulée par la communication cellule-cellule, il est probable que ce sera un outil très utile pour l'analyse des ensembles de données à l'avenir. Il peut être particulièrement efficace pour explorer les cibles médicamenteuses et dans les cas où l'arrangement cellulaire entraîne des modifications de l'expression des gènes. Méthodes mathématiques d'analyse des données transcriptomiques unicellulaires