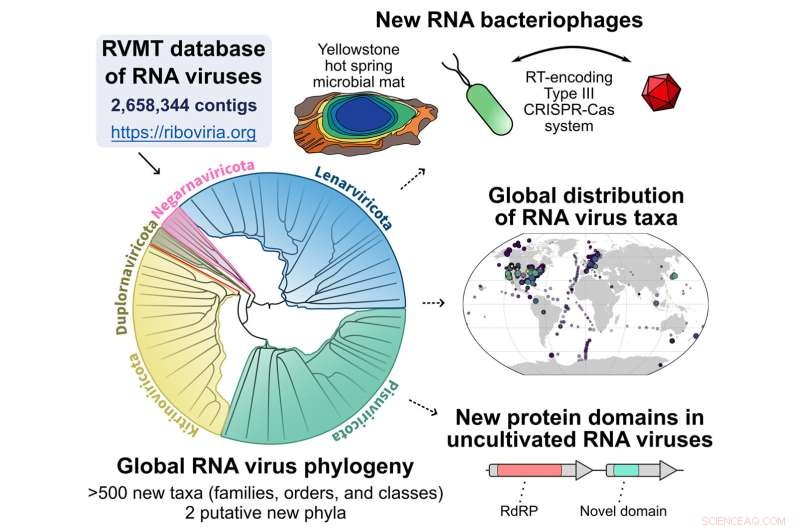
Vue d'ensemble graphique du pipeline commençant par la base de données RNA Virus MetaTranscriptomes (RVMT) pour découvrir l'expansion de la diversité des virus à ARN. Crédit :Simon Roux
Un zoo offrait autrefois un livre de coloriage représentant des ours polaires dans des scènes d'hiver, accompagné de crayons de différentes nuances de blanc. Pour les chercheurs qui recherchent des séquences de virus à ARN dans de grands ensembles de données, leur travail peut s'apparenter à trouver un seul flocon de neige sur une page colorée de ce livre.
Publié en ligne le 28 septembre 2022 dans Cellulaire , une équipe dirigée par des chercheurs de l'Université de Tel Aviv en Israël, du National Center for Biotechnology Information et du Joint Genome Institute (JGI) du Département américain de l'énergie (DOE), une installation utilisateur du Bureau des sciences du DOE située au Lawrence Berkeley National Laboratory ( Berkeley Lab) décrivent un pipeline de calcul qui peut rechercher spécifiquement ces flocons de neige ou séquences de virus à ARN. À l'aide de ce flux de travail, l'équipe a passé au peigne fin plus de 5 000 ensembles de données de séquences d'ARN (métatranscriptomes) générées à partir de divers échantillons environnementaux dans le monde entier, ce qui a multiplié par cinq la diversité des virus à ARN.
"Le monde des virus qui nous entourent est vaste, et nous avons maintenant les moyens de l'explorer", a déclaré Eugene Koonin, chercheur principal au NCBI et l'un des auteurs principaux de l'article, sur la diversité virale découverte. "Bien que les défis techniques de l'analyse des données à cette échelle soient formidables."
Tamis informatiques pour filtrer les séquences
Il y a plus de microbes sur la planète que de particules dans une poignée de terre, et les virus sont largement plus nombreux que les microbes. Les progrès des technologies de séquençage et des outils informatiques ont permis de découvrir une diversité de virus qui infectent non seulement les cultures, les animaux et les humains, mais également les microbes dont la présence ou l'absence peut avoir un impact sur les cycles des nutriments de la planète.
Alors que la plupart des informations génétiques de l'organisme sont codées dans l'ADN, l'ARN fournissant les instructions à l'intérieur de l'ADN à la cellule, les virus à ARN stockent leurs informations génétiques dans l'ARN sans étape d'ADN. "Je dirais que les virus à ARN dans le monde sont encore moins connus que les virus à ADN", a déclaré Simon Roux, un scientifique du JGI et l'un des co-responsables du projet. "Mais comme les virus à ADN, les virus à ARN infectent les microbes partout dans le monde et entraînent la mort cellulaire et/ou des changements profonds dans la physiologie cellulaire pendant l'infection."
Alors que tous les virus à ARN ont un gène qui code pour une enzyme appelée ARN polymérase dirigée par le RNS (RdRP), nécessaire à la réplication de la réplication du génome de l'ARN, sa détection a été un défi. Pour trouver les flocons de neige du virus à ARN dans la tempête de données génomiques, il a fallu développer des tamis informatiques spéciaux pour filtrer les séquences qui étaient peu susceptibles de contenir la séquence RdRP.
Les travaux résultent d'une collaboration à trois qui a débuté en 2019, a rappelé Uri Neri de l'Université de Tel Aviv, l'un des co-responsables du projet et premier auteur de l'étude. Les membres des équipes de Tel Aviv et du NCBI, qui travaillaient déjà ensemble sur l'extraction de virus procaryotes, ont appris de Nikos Kyrpides de JGI que son groupe Microbiome Data Science travaillait également sur l'extraction de virus à ARN. Après quelques réunions virtuelles des trois équipes, il était clair qu'un effort de collaboration plus important serait beaucoup plus efficace pour obtenir des résultats de meilleure qualité par rapport à des efforts individuels plus restreints. C'est également le type d'esprit communautaire synergique et collaboratif que le JGI préconise et promeut activement.
L'équipe a utilisé tous les ensembles de données de métatranscriptome accessibles au public du système Integrated Microbial Genomes &Microbiomes (IMG/M) du JGI. "Nous avons ensuite examiné de nombreux autres échantillons et affiné notre méthodologie", a déclaré Neri. "Notre équipe s'est agrandie, tout comme la portée du projet." À cette fin, a souligné Kyrpides, les contributions des nombreux utilisateurs scientifiques du JGI dans la collecte et la soumission de leurs échantillons de microbiome pour le séquençage au JGI ne peuvent être surestimées. Leur coopération et leur soutien, a-t-il dit, et dans plusieurs cas, leur permission d'utiliser des données de séquence encore non publiées, ont été absolument essentiels pour le succès de cet effort, tout comme la reconnaissance de leur contribution.
Roux et Koonin ont tous deux noté que la pléthore de séquences de virus à ARN découvertes "modifie considérablement la vision globale de la diversité des virus", mais pas aux classifications de niveau supérieur des groupes de virus (phyla.). Les nouvelles séquences comblent certaines lacunes sur les virus existants. groupes tout en ajoutant de nouvelles branches. De plus, les virus à ARN ne semblent pas être répartis uniformément dans le monde.
Un groupe élargi est celui des virus associés aux bactéries; jusqu'à présent, la plupart des virus à ARN connus étaient associés à des eucaryotes. Parallèlement à l'expansion des virus à ARN associés aux bactéries, on a découvert que "quelques bactéries utilisent CRISPR pour se défendre contre l'ARN", a noté Roux, "bien qu'on ne sache pas pourquoi cela est si rarement détecté".
Développer des approches pour réconcilier le "vrai" Big Data
Pour l'équipe, le travail informatique qui a conduit à l'abondance découverte de virus à ARN n'est que le début. "Je dis souvent que le simple fait d'identifier une séquence comme virale n'est même pas la moitié de l'histoire." dit Néri. "Nous avons investi beaucoup d'efforts dans les analyses post-découverte - du mieux que nous pouvions, nous avons essayé de décrire les domaines protéiques que chaque virus porte et qui est leur hôte probable. Nous avons rendu toutes ces informations entièrement gratuites et ouvertement à la disposition de la communauté scientifique au sens large."
Uri Gophna de l'Université de Tel Aviv et Koonin ont tous deux noté que d'autres recherches en parallèle ont signalé des "expansions spectaculaires" similaires du virome à ARN mondial. "Nous devons maintenant comparer et réconcilier les résultats, en proposant un ensemble de données unique et non redondant", a déclaré Koonin. « Espérons que, relativement bientôt, nous pourrons estimer la taille réelle du virome à ARN. Cependant, il s'agit désormais de véritables mégadonnées, nous avons affaire à des milliards de séquences, et bientôt à des billions. Le développement d'approches efficaces et automatisées pour analyser et classer les données de séquence à cette échelle est essentiel." Un outil automatisé pour évaluer la qualité des données virales