La synthèse ou l'étude de certains matériaux en laboratoire pose souvent des défis en raison de problèmes de sécurité, de conditions expérimentales peu pratiques ou de contraintes de coûts. En réponse, les scientifiques se tournent de plus en plus vers des méthodes d'apprentissage profond qui impliquent le développement et l'entraînement de modèles d'apprentissage automatique pour reconnaître des modèles et des relations dans les données qui incluent des informations sur les propriétés, les compositions et les comportements des matériaux.
Grâce à l'apprentissage profond, les scientifiques peuvent rapidement faire des prédictions sur les propriétés des matériaux en fonction de leur composition, de leur structure et d'autres caractéristiques pertinentes, identifier des candidats potentiels pour des recherches plus approfondies et optimiser les conditions de synthèse.
Maintenant, dans une étude parue dans International Union of Crystallography Journal (IUCrJ) , le professeur Takashiro Akitsu, le professeur adjoint Daisuke Nakane et M. Yuji Takiguchi de l'Université des sciences de Tokyo (TUS) ont utilisé l'apprentissage profond pour prédire les aimants monomoléculaires (SMM) à partir d'un pool de 20 000 complexes métalliques. Cette stratégie innovante rationalise le processus de découverte de matériaux en minimisant le besoin de longues expériences.
Les aimants monomoléculaires (SMM) sont des complexes métalliques qui démontrent un comportement de relaxation magnétique au niveau de chaque molécule, où les moments magnétiques subissent des changements ou une relaxation au fil du temps. Ces matériaux ont des applications potentielles dans le développement de mémoires haute densité, de dispositifs spintroniques moléculaires quantiques et de dispositifs informatiques quantiques. Les SMM se caractérisent par une barrière énergétique efficace élevée (Ueff ) pour que le moment magnétique s'inverse. Cependant, ces valeurs sont généralement comprises entre des dizaines et des centaines de Kelvins, ce qui rend les SMM difficiles à synthétiser.
Les chercheurs ont utilisé l'apprentissage profond pour identifier la relation entre les structures moléculaires et le comportement du SMM dans les complexes métalliques avec des ligands de type salen. Ces complexes métalliques ont été choisis car ils peuvent être facilement synthétisés en complexant des aldéhydes et des amines avec divers métaux 3d et 4f.
Pour l'ensemble de données, les chercheurs ont travaillé intensivement pour examiner 800 articles de 2011 à 2021, collectant des informations sur la structure cristalline et déterminant si ces complexes présentaient un comportement SMM. De plus, ils ont obtenu des détails structurels 3D des molécules à partir de la base de données structurelle de Cambridge.
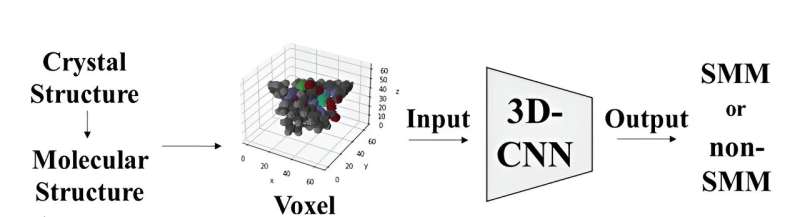
La structure moléculaire des complexes a été représentée à l’aide de voxels ou de pixels 3D, où chaque élément s’est vu attribuer une valeur RVB unique. Par la suite, ces représentations de voxels ont servi d'entrée à un modèle de réseau neuronal convolutif 3D basé sur l'architecture ResNet. Ce modèle a été spécifiquement conçu pour classer les molécules comme SMM ou non-SMM en analysant leurs images moléculaires 3D.
Lorsque le modèle a été formé sur un ensemble de données de structures cristallines de complexes métalliques contenant des complexes de type salen, il a atteint un taux de précision de 70 % pour distinguer les deux catégories. Lorsque le modèle a été testé sur 20 000 structures cristallines de complexes métalliques contenant des bases de Schiff, il a réussi à découvrir les complexes métalliques signalés comme des aimants à molécule unique.
"Il s'agit du premier rapport d'apprentissage profond sur les structures moléculaires des SMM", déclare le professeur Akitsu.
La plupart des structures SMM prédites impliquaient des complexes multinucléaires de dysprosium, connus pour leur Ueff élevé. valeurs. Bien que cette méthode simplifie le processus de découverte du SMM, il est important de noter que les prédictions du modèle sont uniquement basées sur des données d'entraînement et ne lient pas explicitement les structures chimiques à leurs calculs de chimie quantique, une méthode privilégiée dans la conception moléculaire assistée par l'IA. Des recherches expérimentales supplémentaires sont nécessaires pour obtenir les données sur le comportement du SMM dans des conditions uniformes.
Toutefois, cette approche simplifiée a ses avantages. Cela réduit le besoin de calculs informatiques complexes et évite la tâche difficile de simuler le magnétisme.
Le professeur Akitsu conclut :"L'adoption d'une telle approche peut guider la conception de molécules innovantes, entraînant des économies significatives en termes de temps, de ressources et de coûts dans le développement de matériaux fonctionnels."
Plus d'informations : Yuji Takiguchi et al, La prédiction des propriétés magnétiques d'une molécule unique via l'apprentissage profond, IUCrJ (2024). DOI : 10.1107/S2052252524000770
Fourni par l'Université des sciences de Tokyo