De gauche à droite, les chercheurs de St. Jude Timothy Stachowski, Ph.D., du Département de biologie chimique et thérapeutique et Marcus Fischer, Ph.D., Département de biologie chimique et thérapeutique et biologie structurale. Crédit :avec l'aimable autorisation de l'hôpital de recherche pour enfants St. Jude
Les structures protéiques cryogéniques (congelées) sont essentielles pour comprendre la fonction et développer des médicaments. Des scientifiques de l'hôpital de recherche pour enfants St. Jude ont créé un algorithme pour révéler que la congélation des protéines peut créer des "artefacts", des erreurs qui provoquent des résultats trompeurs. La recherche est parue récemment dans Angewandte Chemie International Edition et a souligné l'importance des réseaux d'eau dans les interactions protéine-ligand. Les résultats remettent en question l'opinion commune selon laquelle les positions de l'eau cryogénique bien résolues sont à la fois précises et exactes.
Les ligands sont des molécules qui se lient à une protéine réceptrice. Lorsqu'un ligand se lie à une protéine, la conformation (forme) peut changer, initiant différents types d'activité dans la cellule. La liaison protéine-ligand et les changements de forme qui en résultent sont des éléments cruciaux à prendre en compte lors des efforts de développement de médicaments.
"Si vous ne regardez que les données cryogéniques, les informations utilisées pour la découverte de médicaments contiennent des artefacts dont vous ne sauriez pas qu'ils étaient là", a déclaré l'auteur correspondant Marcus Fischer, Ph.D., St. Jude Departments of Chemical Biology and Thérapeutique et biologie structurale. "Nous avons développé un moyen de démêler ces artefacts. En utilisant des comparaisons appariées entre les températures cryogéniques et ambiantes, vous pouvez identifier les parties de la protéine qui sont affectées par la température."
Les chercheurs utilisent souvent les structures protéiques disponibles en extrayant les informations d'une base de données appelée Research Collaboratory for Structural Bioinformatics Protein Data Bank. Environ 95 % de ces structures sont capturées par cryogénie, puis modélisées dans la base de données pour en faciliter l'utilisation. Les découvreurs de médicaments regardent rarement de près les données expérimentales brutes, qui se présentent sous la forme d'une carte de densité électronique. L'interrogation de cartes plutôt que de modèles structurels offre une approche impartiale pour révéler des caractéristiques dynamiques et des artefacts cryogéniques.
L'algorithme Flipper met en évidence les changements importants
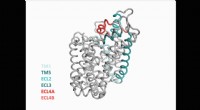
Fischer et son équipe ont développé un algorithme, appelé Flipper, qui examine les données expérimentales brutes dans les cartes de densité électronique. Flipper identifie les pics de carte (signaux) qui seraient autrement invisibles. Ces pics correspondent aux parties de protéines de résidus spécifiques qui ont des conformations sensibles à la température. Ces résidus peuvent modifier la préférence relative d'un état par rapport à un autre, ou "basculer" dans leur densité, se déplaçant entre les conformations, d'où l'algorithme tire son nom.
Les chercheurs ont utilisé cette approche pour identifier les résidus qui réagissent aux changements de température et pour suivre les résidus dans un système semblable à un code-barres sur l'ensemble de la protéine. Cela a permis aux scientifiques de voir comment les résidus à l'intérieur et à l'extérieur du site de liaison du ligand réagissent aux températures de congélation ou de réchauffement.
"Avec Flipper, nous pouvons détecter des changements mineurs mais importants dans les structures des protéines à partir de la température ou d'autres facteurs", a déclaré le premier auteur Timothy Stachowski, Ph.D., St. Jude Chemical Biology and Therapeutics. "Il est important de corriger ces détails dès le début du processus de découverte de médicaments, sinon les efforts de recherche pourraient être égarés."
Étant donné que les effets de la température et du réseau hydrique influencent un grand nombre de structures, les résultats peuvent avoir un impact généralisé sur le développement de médicaments.
Une nouvelle appréciation pour les réseaux d'eau
Forts de leur nouvelle approche, les chercheurs ont mené une analyse systématique montrant l'importance des réseaux d'eau. L'eau, l'une des molécules les plus cruciales et les plus abondantes sur Terre, joue un rôle actif dans le processus de congélation des conformations. Cela est particulièrement vrai au niveau des sites de liaison protéine-ligand.
"C'est la première fois que nous montrons systématiquement l'importance de la température sur les réseaux d'eau pour moduler l'interface de liaison du ligand, où se produit la biologie", a déclaré Fischer. "L'eau est souvent ignorée dans le processus de découverte de médicaments, mais nous avons montré qu'en plus d'avoir un effet profond sur la liaison du ligand, l'eau influence également les résidus du site de liaison, les capturant dans des positions qui diffèrent en fonction de la température."
Flipper et le système de code-barres conformationnel qui facilite les comparaisons de différents ligands à différentes températures sont disponibles gratuitement pour permettre à d'autres chercheurs d'identifier de tels modèles dans leurs propres ensembles de données. La comparaison des structures cryogéniques avec des échantillons à température ambiante peut aider à identifier les erreurs dans les modèles de calcul