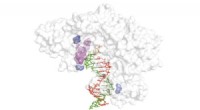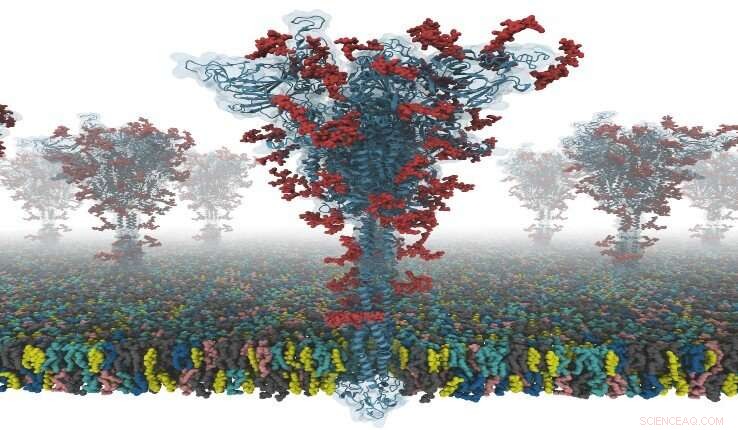
Un modèle d'une protéine S. Crédit :Dr Yeolkyo Choi/Lehigh
Le virus SARS coronavirus 2 (SARS-CoV-2) est la cause connue de la maladie à coronavirus 2019 (COVID-19). Le « pic » ou protéine S facilite l'entrée virale dans les cellules hôtes.
Aujourd'hui, un groupe de chercheurs de l'Université nationale de Séoul en Corée du Sud, Université de Cambridge au Royaume-Uni, et l'Université Lehigh aux États-Unis, ont travaillé ensemble pour produire les premiers modèles open source tout-atome d'une protéine S pleine longueur. Les chercheurs disent que cela est particulièrement important car la protéine S joue un rôle central dans l'entrée virale dans les cellules, ce qui en fait une cible principale pour le développement de vaccins et de médicaments antiviraux.
Les détails peuvent être trouvés dans un document , "Développement d'un modèle de protéine de pointe SARS-CoV-2 pleine longueur entièrement glycosylée dans une membrane virale" vient d'être publié en ligne dans Le Journal de Chimie Physique B .
Cette démo vidéo illustre comment construire ce système membranaire à partir de leurs modèles de protéines SARS-CoV-2 S. Le programme de modélisme est en accès libre et peut être consulté depuis la page d'accueil de CHARMM-GUI en cliquant sur le lien Archive COVID-19 , ou en cliquant sur le lien d'archive dans l'en-tête, puis le lien Protéines COVID-19 dans la barre latérale gauche.
Développé par Wonpil Im, professeur au Département des sciences biologiques et du Département de bio-ingénierie de l'Université Lehigh, CHARMM-GUI (GUI =graphical user interface) est un programme qui simule simplement des systèmes biomoléculaires complexes, précisément et rapidement. Im le décrit comme un "microscope informatique" qui permet aux scientifiques de comprendre les interactions au niveau moléculaire qui ne peuvent être observées autrement. Vous trouverez plus d'informations sur CHARMM-GUI dans cette vidéo.
"Nos modèles sont les premiers modèles de protéines de pointe (S) SARS-CoV-2 pleine longueur entièrement glycosylés qui sont disponibles pour d'autres scientifiques, " dit Im. " J'ai eu la chance de collaborer avec le Dr Chaok Seok de l'Université nationale de Séoul en Corée et le Dr Tristan Croll de l'Université de Cambridge au Royaume-Uni. Notre équipe a passé des jours et des nuits à construire ces modèles très soigneusement à partir du cryo- Parties de la structure EM. La modélisation était très difficile car il y avait de nombreuses régions où la modélisation simple ne parvenait pas à fournir des modèles de haute qualité. »
Les scientifiques peuvent utiliser les modèles pour mener des recherches de simulation innovantes et novatrices pour la prévention et le traitement du COVID-19, selon Im.
La structure de la protéine S a été déterminée par cryo-EM avec le RBD up (PDB ID :6VSB), et avec le RBD vers le bas (ID PDB :6VXX). Mais, ce modèle a de nombreux résidus manquants. Donc, ils ont d'abord modélisé les résidus d'acides aminés manquants, puis d'autres domaines manquants. En outre, ils ont modélisé tous les glycanes (ou glucides) potentiels attachés à la protéine S. Ces glycanes empêchent la reconnaissance des anticorps, ce qui rend difficile le développement d'un vaccin. Ils ont également construit un système de membrane virale d'une protéine S pour la simulation de la dynamique moléculaire.