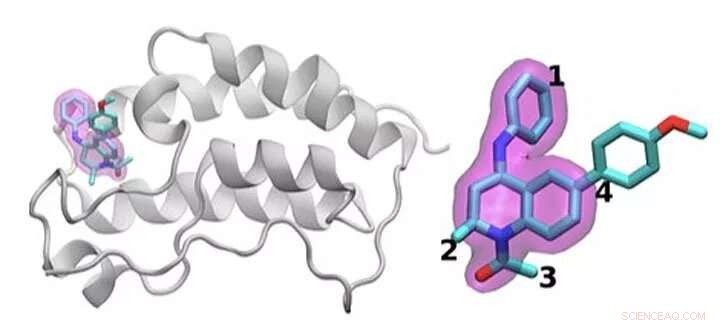
Un schéma de la protéine BRD4 liée à l'un des 16 médicaments basés sur le même échafaudage de tétrahydroquinoléine (mis en évidence en magenta). Les régions qui sont chimiquement modifiées entre les médicaments étudiés dans cette étude sont étiquetées de 1 à 4. En règle générale, seule une petite modification est apportée à la structure chimique d'un médicament à l'autre. Cette approche conservatrice permet aux chercheurs d'explorer pourquoi un médicament est efficace alors qu'un autre ne l'est pas. Crédit :Laboratoire national de Brookhaven
Identifier le traitement médicamenteux optimal, c'est comme toucher une cible mouvante. Pour arrêter la maladie, les médicaments à petites molécules se lient étroitement à une protéine importante, bloquant ses effets dans le corps. Même les médicaments approuvés ne fonctionnent généralement pas chez tous les patients. Et avec le temps, les agents infectieux ou les cellules cancéreuses peuvent muter, rendant inutile un médicament autrefois efficace.
Un problème physique central sous-tend tous ces enjeux :optimiser l'interaction entre la molécule médicamenteuse et sa cible protéique. Les variations des molécules candidates-médicaments, la gamme de mutations dans les protéines et la complexité globale de ces interactions physiques rendent ce travail difficile.
Shantenu Jha du Laboratoire national de Brookhaven du Département de l'énergie (DOE) et de l'Université Rutgers dirige une équipe qui tente de rationaliser les méthodes de calcul afin que les superordinateurs puissent assumer une partie de cette immense charge de travail. Ils ont trouvé une nouvelle stratégie pour s'attaquer à une partie :différencier la façon dont les candidats-médicaments interagissent et se lient à une protéine ciblée.
Pour leur travail, Jha et ses collègues ont remporté l'année dernière le prix IEEE International Scalable Computing Challenge (SCALE), qui reconnaît des solutions informatiques évolutives aux problèmes scientifiques et techniques du monde réel.
Pour concevoir un nouveau médicament, une société pharmaceutique peut commencer avec une bibliothèque de millions de molécules candidates qu'elle réduit à des milliers qui montrent une liaison initiale à une protéine cible. Le raffinement de ces options en un médicament utile pouvant être testé chez l'homme peut impliquer des expériences approfondies pour ajouter ou soustraire des groupes d'atomes à des emplacements clés de la molécule et tester comment chacun de ces changements modifie la façon dont la petite molécule et la protéine interagissent.
Les simulations peuvent aider dans ce processus. Plus grand, des superordinateurs plus rapides et des algorithmes de plus en plus sophistiqués peuvent incorporer une physique réaliste et calculer les énergies de liaison entre diverses petites molécules et protéines. De telles méthodes peuvent consommer des ressources de calcul importantes, cependant, pour atteindre la précision nécessaire. Les simulations utiles à l'industrie doivent également fournir des réponses rapides. En raison du tiraillement entre la précision et la vitesse, les chercheurs innovent en permanence, développer des algorithmes plus efficaces et améliorer les performances, dit Jha.
Ce problème nécessite également de gérer les ressources de calcul différemment que pour de nombreux autres problèmes à grande échelle. Au lieu de concevoir une simulation unique qui évolue pour utiliser un supercalculateur entier, les chercheurs exécutent simultanément de nombreux modèles plus petits qui façonnent les uns les autres et la trajectoire des calculs futurs, une stratégie connue sous le nom de calcul d'ensemble, ou des flux de travail complexes.
"Pensez à cela comme essayer d'explorer un très grand paysage ouvert pour essayer de trouver où vous pourriez être en mesure d'obtenir le meilleur candidat-médicament, " dit Jha. Dans le passé, des chercheurs ont demandé aux ordinateurs de naviguer dans ce paysage en faisant des choix statistiques aléatoires. À un point de décision, la moitié des calculs pourraient suivre un chemin, l'autre moitié l'autre.
Jha et son équipe cherchent des moyens d'aider ces simulations à apprendre du paysage à la place. Ingérer puis partager des données en temps réel n'est pas facile, Jha dit, « et c'est ce qui a nécessité une partie de l'innovation technologique à faire à grande échelle. » Lui et son équipe basée à Rutgers collaborent avec le groupe de Peter Coveney à l'University College London sur ce travail.
Pour tester cette idée, ils ont utilisé des algorithmes qui prédisent l'affinité de liaison et ont introduit des versions simplifiées dans un cadre HTBAC, pour le calculateur d'affinité de liaison à haut débit. Une telle calculatrice, connu sous le nom d'ESMACS, les aide à éliminer les molécules qui se lient mal à une protéine cible. L'autre, LIENS, est plus précis mais plus limité dans sa portée et nécessite 2,5 fois plus de ressources de calcul. Néanmoins, il peut aider les chercheurs à optimiser une interaction prometteuse entre un médicament et une protéine. Le framework HTBAC les aide à implémenter efficacement ces algorithmes, en sauvegardant l'algorithme le plus intensif pour les situations où il est nécessaire.
L'équipe a démontré l'idée en examinant 16 candidats-médicaments d'une bibliothèque de molécules à GlaxoSmithKline (GSK) avec leur cible, BRD4-BD1—une protéine importante dans le cancer du sein et les maladies inflammatoires. Les candidats-médicaments avaient la même structure centrale mais différaient dans quatre zones distinctes autour des bords de la molécule.
Dans cette étude initiale, l'équipe a exécuté des milliers de processus simultanément sur 32, 000 carottes sur Blue Waters, un superordinateur de la National Science Foundation (NSF) de l'Université de l'Illinois à Urbana-Champaign. Ils ont effectué des calculs similaires sur Titan, le supercalculateur Cray XK7 de l'Oak Ridge Leadership Computing Facility, une installation utilisateur du DOE Office of Science. L'équipe a réussi à distinguer la liaison de ces 16 candidats médicaments, la plus grande simulation de ce type à ce jour. "Nous n'avons pas seulement atteint une échelle sans précédent, " dit Jha. "Notre approche montre la capacité de se différencier."
Ils ont remporté leur prix SCALE pour cette première preuve de concept. Le défi maintenant, Jha dit, s'assure que cela ne fonctionne pas seulement pour BRD4, mais aussi pour d'autres combinaisons de molécules médicamenteuses et de cibles protéiques.
Si les chercheurs peuvent continuer à élargir leur approche, de telles techniques pourraient éventuellement aider à accélérer la découverte de médicaments et permettre une médecine personnalisée. Mais pour examiner des problèmes plus réalistes, ils auront besoin de plus de puissance de calcul. "Nous sommes au milieu de cette tension entre un très grand espace chimique que nous, en principe, besoin d'explorer, et, malheureusement des ressources informatiques limitées." dit Jha.
Même si le calcul intensif s'étend vers l'exascale, les informaticiens peuvent plus que combler le vide en ajoutant une physique plus réaliste à leurs modèles. Dans un avenir prévisible, les chercheurs devront faire preuve d'ingéniosité pour étendre ces calculs. La nécessité est la mère de l'innovation, Jha dit, précisément parce que la science moléculaire n'aura pas la quantité idéale de ressources informatiques pour effectuer des simulations.
Mais l'informatique exascale peut les aider à se rapprocher de leurs objectifs. En plus de travailler avec University College London et GSK, Jha et ses collègues collaborent avec Rick Stevens du Laboratoire national d'Argonne et l'équipe CANcer Distributed Learning Environment (CANDLE). Ce projet de co-conception au sein du projet d'informatique exascale du DOE construit des réseaux de neurones profonds et des techniques générales d'apprentissage automatique pour étudier le cancer. Les algorithmes et les logiciels de HTBAC pourraient compléter l'accent mis par CANDLE sur ces approches.
Cette collaboration plus large entre le groupe de Jha, l'équipe CANDLE et le laboratoire de John Chodera au Memorial Sloan-Kettering Cancer Center ont mené au projet INSPIRE (Integrated and Scalable Prediction of Resistance). Cette équipe a déjà effectué des simulations sur le supercalculateur Summit du DOE au Oak Ridge National Laboratory. Il poursuivra bientôt ce travail sur Frontera, la machine de leadership de la NSF à l'Université du Texas au Texas Advanced Computing Center d'Austin.
"Nous avons soif de progrès et d'améliorations méthodologiques plus importants, " dit Jha. "Nous aimerions voir comment ces approches assez complémentaires pourraient fonctionner de manière intégrative vers cette grande vision."