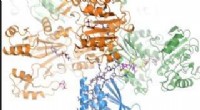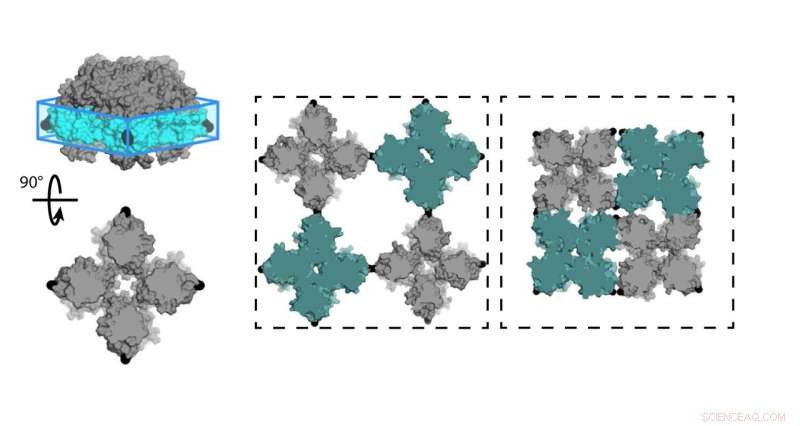
Chimistes de l'Université de Californie, San Diego (UCSD) a conçu une feuille de protéines (C98RhuA) qui bascule entre différents états de porosité et de densité. Les cellules du réseau cristallin sont articulées aux coins du tétramère C98RhuA, lui permettant de tourner et d'ouvrir ou de fermer le pore. Crédit :Robert Alberstein et al.
Qu'est-ce qui fait que le kevlar arrête une balle, au niveau atomique ?
Les propriétés des matériaux ressortent de leur structure moléculaire ou atomique, pourtant, de nombreux détails entre le micro et le macro restent un mystère pour la science. Les scientifiques recherchent activement la conception rationnelle d'architectures supramoléculaires ciblées, dans le but de concevoir leur dynamique structurelle et leur réponse aux signaux environnementaux.
Une équipe de chimistes de l'Université de Californie, San Diego (UCSD) a maintenant conçu un cristal de protéine bidimensionnel qui bascule entre des états de porosité et de densité variables. Il s'agit d'une première dans la conception biomoléculaire qui combine des études expérimentales avec des calculs effectués sur des superordinateurs. La recherche, publié en avril 2018 dans Chimie de la nature , pourrait aider à créer de nouveaux matériaux pour les énergies renouvelables, Médicament, purification de l'eau, et plus.
"Nous avons fait un ensemble complet de simulations et d'expériences de dynamique moléculaire, qui expliquait la base de la dynamique structurelle inhabituelle de ces protéines artificielles, sur la base desquelles nous avons pu prendre des décisions rationnelles et modifier la dynamique structurelle de l'ensemble, " a déclaré le co-auteur de l'étude Akif Tezcan, professeur de chimie et de biochimie à l'UCSD.
L'équipe de Tezcan a travaillé avec la protéine L-rhamnulose-1-phosphate aldolase (RhuA), qui a été modifié avec des acides aminés de cystéine dans ses quatre coins à la position 98 (C98RhuA). Lui et son groupe avaient déjà publié des travaux sur l'auto-assemblage de cet artificiel, architecture protéique bidimensionnelle, qui, selon lui, montrait un comportement intéressant appelé auxéticité.
"Ces assemblages cristallins peuvent effectivement s'ouvrir et se fermer en cohérence, " dit Tezcan. " Comme ils le font, ils rétrécissent ou s'étendent également dans les directions X et Y, ce qui est le contraire de ce que font les matériaux normaux. Nous voulions étudier à quoi ces mouvements sont dus et ce qui les régit. une balle jouet qui se dilate à travers ses charnières en forme de ciseaux lorsque vous séparez les extrémités.
"Notre objectif était de pouvoir faire la même chose, utiliser des protéines comme éléments constitutifs, créer de nouveaux types de matériaux aux propriétés avancées, " a déclaré Tezcan. " L'exemple que nous étudions ici était essentiellement le fruit de ces efforts, où nous avons utilisé cette protéine particulière qui a une forme carrée, que nous attachions les uns aux autres par des liaisons chimiques qui étaient réversibles et agissaient comme des charnières. Cela a permis à ces matériaux de former des cristaux très bien ordonnés qui étaient également dynamiques en raison de la flexibilité de ces liaisons chimiques, qui a fini par nous donner ces nouvelles, propriétés émergentes."
Le contrôle de l'ouverture et de la fermeture des pores dans les réseaux 2-D de la protéine C98RhuA pourrait capturer ou libérer des cibles moléculaires spécifiques utiles pour l'administration de médicaments ou la création de meilleures batteries avec plus de recherche, dit Tezcan. Ou ils pourraient sélectivement traverser ou bloquer le passage de molécules biologiques et filtrer l'eau.

Le supercalculateur Maverick est une ressource dédiée à la visualisation et à l'analyse de données du Texas Advanced Computing Center qui est architecturée avec 132 unités de traitement graphique (GPU) NVIDIA Tesla K40 'Atlas' pour la visualisation à distance et le calcul GPU à la communauté nationale. Crédit :TACC
« Notre idée était de pouvoir construire des matériaux complexes, comme l'évolution l'a fait, utiliser des protéines comme éléments constitutifs, " a déclaré Tezcan.
La façon dont l'équipe de Tezcan l'a fait était d'abord d'exprimer les protéines dans les cellules de la bactérie E. coli et de les purifier, après quoi ils ont induit la formation des liaisons chimiques qui créent réellement les cristaux de C98RhuA, qui varient en fonction de leur état d'oxydation, par l'ajout de produits chimiques redox-actifs.
"Une fois les cristaux formés, la grande caractérisation devient l'ouverture ou la proximité des cristaux eux-mêmes, " expliqua Tezcan, qui a été déterminé par l'analyse statistique de centaines d'images capturées par microscopie électronique.
Les expériences ont fonctionné de pair avec le calcul, principalement des simulations tout-atome utilisant le logiciel NAMD développé à l'Université de l'Illinois à Urbana Champaign par le groupe du regretté biophysicien Klaus Schulten.
L'équipe de Tezcan a utilisé un système réduit de seulement quatre protéines liées entre elles, qui peut être carrelé à l'infini pour aller au fond de la façon dont le cristal s'ouvre et se ferme. « Le système réduit nous a permis de rendre ces calculs réalisables pour nous, car il y a encore des centaines de milliers d'atomes, même dans ce système réduit, " a déclaré Tezcan. Son équipe a profité de fonctionnalités spécifiques au C98RhuA, comme l'utilisation d'une seule coordonnée de réaction correspondant à son ouverture. "Nous avons vraiment pu valider ce modèle comme étant représentatif de ce que nous avons observé dans l'expérimentation, " a déclaré Tezcan.
Les simulations moléculaires de tous les atomes des réseaux cristallins C98RhuA ont été utilisées pour cartographier le paysage de l'énergie libre. Ce paysage énergétique ressemble à un paysage naturel, avec des vallées, montagnes, et cols de montagne, a expliqué le co-auteur de l'étude Francesco Paesani, professeur de chimie et de biochimie à l'UCSD.
"Les vallées deviennent les configurations les plus stables de vos assemblages de protéines, " Paesani a dit, que le système moléculaire préfère à avoir à dépenser de l'énergie pour franchir une montagne. Et les cols de montagne montrent le chemin d'une structure stable à une autre.
"Typiquement, les calculs d'énergie libre sont très coûteux et difficiles, car essentiellement ce que vous essayez de faire est d'échantillonner toutes les configurations possibles d'un système moléculaire qui contient des milliers d'atomes. Et vous voulez savoir combien de positions ces atomes peuvent acquérir lors d'une simulation. Cela prend beaucoup de temps et beaucoup de ressources informatiques, " a déclaré Paesani.
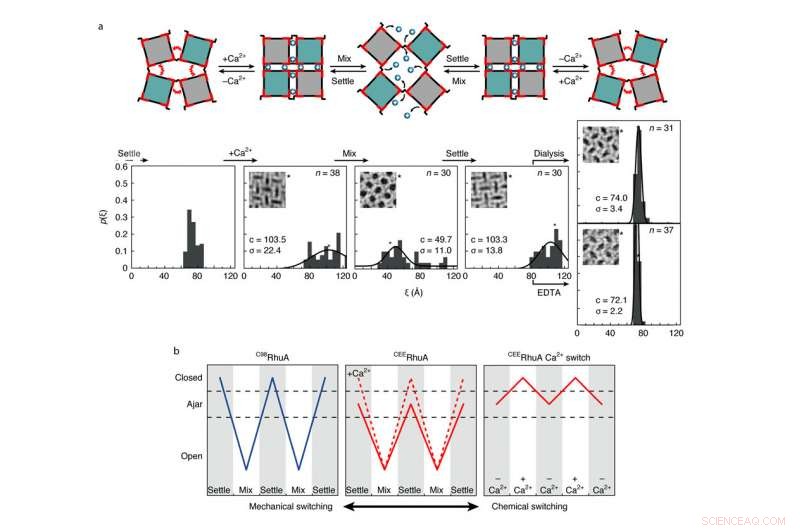
Comportement de commutation chimique et mécanique des cristaux CEERhuA. une, En haut :schéma illustrant tous les modes de commutation possibles des réseaux CEERhuA. En bas :distribution(s) expérimentale(s) correspondant aux états directement au-dessus. L'ajout de 20 mM de Ca2+ à la population d'équilibre « entrouverte » de cristaux CEERhuA a induit un changement vers des conformations plus fermées, à partir de laquelle une commutation mécanique de type C98RhuA était possible. La conformation entrouverte était entièrement récupérable lors de l'élimination du Ca2+ par dialyse ou EDTA, fournissant ainsi trois modes de commutation distincts. Les ajustements gaussiens à chaque distribution sont étiquetés avec leur centre (c) et s.d. (σ). n est le nombre de cristaux analysés. La conformation en treillis de chaque encart est marquée d'un astérisque. b, Résumé des modes de commutation pour les cristaux RhuA. Contrairement à C98RhuA, CEERhuA a deux modes mécaniques dictés par la présence de Ca2+, ainsi qu'un mode purement chimique via l'ajout ou l'élimination de Ca2+. Crédit :Robert Alberstein et al.
Pour relever ces défis et d'autres encore, Paesani a obtenu des allocations de supercalculateurs via XSEDE, l'environnement de découverte des sciences et de l'ingénierie extrêmes, financé par la National Science Foundation.
"Heureusement, XSEDE nous a fourni une allocation sur Maverick, les clusters de calcul GPU du Texas Advanced Computing Center (TACC), " a déclaré Paesani. Maverick est une ressource dédiée à la visualisation et à l'analyse de données architecturée avec 132 unités de traitement graphique (GPU) NVIDIA Tesla K40 "Atlas" pour la visualisation à distance et le calcul GPU à la communauté nationale.
"Cela nous a été très utile, car le logiciel NAMD que nous utilisons fonctionne très bien sur les GPU. Cela nous permet d'accélérer les calculs par ordre de grandeur, " a dit Paesani. " Aujourd'hui, nous pouvons nous permettre des calculs auxquels nous ne pouvions même pas rêver il y a dix ans à cause de ces développements, à la fois sur le logiciel NAMD et sur le matériel. Tous ces clusters de calcul fournis par XSEDE sont en fait très utiles pour toutes les simulations de dynamique moléculaire. »
Grâce à XSEDE, Paesani a utilisé plusieurs systèmes de calcul intensif, dont Gordon, Comète, et Trestles au Supercomputer Center de San Diego; Kraken à l'Institut national des sciences informatiques ; et Ranger, Débandade, et Stampede2 au TACC.
« Parce que toutes les simulations ont été exécutées sur des GPU, Maverick était le choix parfait pour ce type d'application, " a déclaré Paesani.
Le calcul et l'expérimentation ont travaillé ensemble pour produire des résultats. "Je pense que c'est un bel exemple de synergie entre théorie et expérimentation, " Paesani a dit. " L'expérience a posé la première question. La théorie et la simulation informatique ont abordé cette question, fournir une certaine compréhension du mécanisme. Et puis nous avons utilisé la simulation informatique pour faire des prédictions et demander aux expériences de tester la validité de ces hypothèses. Tout s'est très bien passé car les simulations ont expliqué les expériences au début. Les prédictions qui ont été faites ont été confirmées par les expériences à la fin. C'est un exemple de la synergie parfaite entre expérimentations et modélisation théorique."
Tezcan a ajouté que « les chimistes aiment traditionnellement construire des molécules complexes à partir de blocs de construction plus simples, et on peut envisager de faire une telle combinaison de conception, expérimenter et calculer des molécules plus petites pour prédire leur comportement. Mais le fait que nous puissions le faire sur des molécules composées de centaines de milliers d'atomes est sans précédent."
L'équipe scientifique a également utilisé des simulations de dynamique moléculaire pour étudier de manière rigoureuse le rôle de l'eau dans la direction du mouvement du réseau de C98RhuA. "Cette étude nous a montré à quel point le rôle actif de l'eau est important dans le contrôle de la dynamique structurelle des macromolécules complexes, qui en biochimie peut être négligé, " dit Tezcan. " Mais cette étude a montré, très clairement, que la dynamique de ces protéines est déterminée activement par la dynamique de l'eau, ce qui, je pense, met l'importance de l'eau au premier plan."
Rob Albertstein, étudiant diplômé du groupe Tezcan et premier auteur de l'article Nature Chemistry, a ajouté "Au cœur de cette recherche se trouve la compréhension de la manière dont les propriétés des matériaux découlent de la structure moléculaire ou atomique sous-jacente. C'est très difficile à décrire. Dans ce cas, nous avons vraiment cherché à établir cette connexion aussi clairement que nous pouvions le comprendre nous-mêmes et vraiment montrer non seulement à partir de l'expérience, où l'on peut regarder le comportement à l'échelle macro de ces matériaux, mais ensuite, avec le calcul, reliez ce comportement à ce qui se passe réellement à l'échelle des molécules. Alors que nous continuons à nous développer en tant que société, nous devons développer de nouveaux matériaux pour de nouveaux types de problèmes mondiaux (purification de l'eau, etc), donc comprendre cette relation entre la structure atomique et la propriété matérielle elle-même et la capacité de prédire celles-ci va devenir de plus en plus importante. »