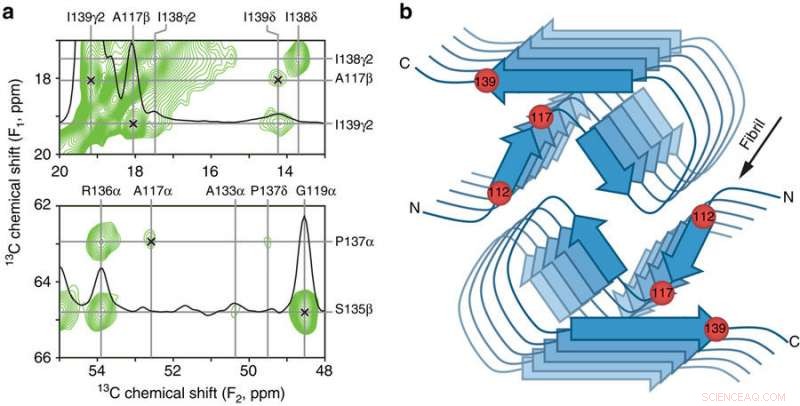
Contacts interrésidus clés et modèle schématique du noyau β-amyloïde PrP23-144 humain. a Petites régions d'un spectre RMN à l'état solide 13C-13C DARR bidimensionnel à 900 ms enregistré avec un temps de mélange de 500 ms pour les fibrilles amyloïdes générées à partir de huPrP23-144 exprimées avec du 3-13C-pyruvate comme source de carbone. Les régions spectrales contiennent les principales contraintes sur la structure du noyau amyloïde [hu] sous la forme de corrélations à longue portée sans ambiguïté (indiquées par des x) entre les atomes 13C suivants :A117Cβ-I139Cγ2, A117Cβ-I139Cδ, A117Cα-P137Cα, et G119Cα-S135Cβ. b Modèle schématique du noyau amyloïde [hu] basé sur la combinaison de données de RMN à l'état solide et de microscopie électronique à transmission à faisceau incliné (voir le texte pour plus de détails). Dans ce modèle, les fibrilles amyloïdes [hu] sont constituées de deux protofilaments dans un arrangement symétrique en C2 avec des régions de feuillets parallèles à l'axe long des fibrilles. Les emplacements approximatifs des résidus d'acides aminés 112, 117, et 139, qui ont un impact majeur sur la structure adoptée par PrP23-144 amyloïde comme discuté dans le texte, sont indiqués par des sphères rouges. Crédit: Communication Nature (2017). DOI :10.1038/s41467-017-00794-z
Des chercheurs qui étudient une protéine qui provoque une maladie cérébrale dégénérative héréditaire chez l'homme ont découvert que l'humain, formes de souris et de hamster de la protéine, qui ont des séquences d'acides aminés presque identiques, présentent des structures tridimensionnelles distinctes au niveau atomique.
La protéine provoque une angiopathie amyloïde cérébrale humaine familiale (AAC), et l'étude, qui apparaît dans Communication Nature , est le premier à examiner les formes de la protéine chez trois espèces différentes.
Christophe Jaroniec, professeur de chimie et de biochimie à l'Ohio State University, a déclaré que les résultats mettent en évidence le fait que des altérations mineures d'acides aminés simples peuvent provoquer de profondes différences de structure et de fonction parmi cette famille de protéines.
"Les différences à grande échelle dans les structures et les caractéristiques de transmission de ces protéines - causées par des différences apparemment insignifiantes dans les positions de quelques atomes de carbone et d'hydrogène - sont assez remarquables, " a déclaré Jaroniec.
L'étude ne constitue pas la base d'un nouveau test ou traitement pour le CAA, mais utilise plutôt ces protéines comme modèles pour comprendre les aspects fondamentaux de la transmission interspécifique de toute une classe de maladies dégénératives du cerveau appelées maladies à prions, il expliqua. Il souligne également l'utilité de la spectroscopie par résonance magnétique nucléaire (RMN) à l'état solide pour l'imagerie des structures des protéines associées aux maladies à prions.
Les chercheurs savent que dans le corps, les molécules de protéines associées au CAA forment des plaques qui se logent dans les parois des vaisseaux sanguins du cerveau, mais il n'y a pas eu d'examens détaillés de la structure moléculaire de ces plaques jusqu'à récemment. En 2008, Des chercheurs de l'État de l'Ohio et leurs partenaires de la Case Western Reserve University ont réalisé les premières études à l'état solide de la variante de protéine prion pertinente, et réduit la liste des acides aminés potentiellement critiques pour sa fonction à environ 30.
Maintenant, ils ont démontré qu'un seul acide aminé, connu par son nombre le long de la chaîne protéique, 139 - est la clé de cette variante de protéine prion adoptant une structure « de type humain » par rapport à une structure « de type hamster », tandis qu'un autre acide aminé, 112, régit les différences structurelles entre les versions humaine et murine de la protéine. Ils ont également montré que ces deux acides aminés semblent être responsables de l'émergence de « souches de prions » structurellement distinctes au sein d'une même séquence protéique, par analogie avec des souches distinctes d'un virus.
Les maladies à prions les plus connues sont l'encéphalopathie spongiforme bovine (souvent appelée « maladie de la vache folle ») et la maladie de Creutzfeldt-Jakob chez l'homme. Tous sont incurables et mortels, et certains peuvent également être transmissibles. On pense que les structures adoptées par les protéines prions du cerveau dans les plaques sont essentielles à leur capacité à se transmettre entre différents hôtes et à provoquer des maladies.
"Notre groupe travaille actuellement à la détermination des structures moléculaires à haute résolution des variants tronqués de la protéine prion associés au CAA humain familial afin d'acquérir une compréhension atomistique complète des facteurs qui sous-tendent leur transmission, et la présente étude est un tremplin majeur dans cet effort, " a déclaré Jaroniec.
"Nous espérons qu'un jour notre groupe et d'autres chercheurs pourront utiliser des méthodologies similaires pour démêler la base structurelle des maladies à prions transmissibles, " il ajouta.