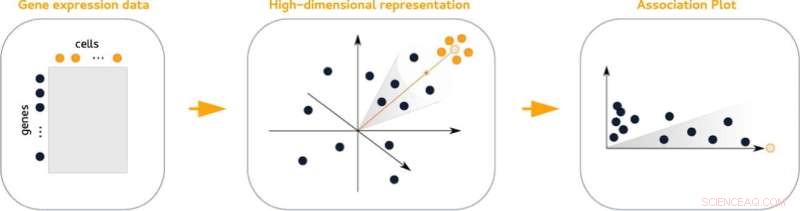
Résumé graphique. Crédit :Journal of Molecular Biology (2022). DOI :10.1016/j.jmb.2022.167525
Quels gènes sont spécifiques à un certain type de cellule, c'est-à-dire « marquent » leur identité ? Avec la taille croissante des ensembles de données de nos jours, répondre à cette question est souvent difficile. Souvent, les gènes marqueurs sont simplement des gènes qui ont été trouvés dans des populations cellulaires spécifiques. Cependant, de nombreux autres gènes pourraient être caractéristiques d'un type cellulaire particulier mais rester à découvrir.
Les diagrammes d'association (APL), une nouvelle méthode statistique de visualisation de l'activité des gènes au sein d'un amas cellulaire, facilitent la recherche de ses gènes marqueurs. Les parcelles comparent l'activité des gènes d'un cluster donné avec tous les autres clusters de l'ensemble de données. De plus, ils permettent de voir facilement quels gènes sont partagés avec d'autres clusters.
« Les diagrammes d'association nous permettent non seulement d'identifier de nouveaux gènes marqueurs. Cela fonctionne également dans l'autre sens :nous sommes capables de faire correspondre des grappes d'identité inconnue dans un ensemble de données à des types de cellules, sur la base d'une liste fournie de gènes marqueurs », explique Elzbieta Gralinska. de l'Institut Max Planck de génétique moléculaire à Berlin.
Le biotechnologue travaille dans l'équipe de Martin Vingron, qui a développé la technique. Les chercheurs ont démontré la fonctionnalité de la technique sur deux ensembles de données accessibles au public et ont publié les résultats dans le Journal of Molecular Biology . De plus, APL a été publié en tant que module gratuit pour l'environnement statistique R. Le package APL permet aux chercheurs d'inspecter visuellement leurs données unicellulaires et de sélectionner des gènes individuels avec le curseur pour en savoir plus sur les détails.
Analyser et regrouper des cellules individuelles
Pourquoi est-il nécessaire d'identifier les gènes marqueurs en premier lieu ? Les technologies de séquençage modernes sont capables de déchiffrer des molécules d'ARN individuelles dans des cellules individuelles. A partir d'un échantillon de sang, par exemple, chaque cellule peut être séparée et un échantillon des ARN de la cellule peut être décodé. Ces données unicellulaires représentent les gènes actifs qui ont été transcrits en molécules d'ARN.
L'avantage :au lieu de se demander à quel type de cellule appartient un ARN particulier, il peut être retracé jusqu'à sa cellule d'origine. L'inconvénient :le séquençage de milliers d'ARN dans chaque cellule parmi des dizaines de milliers de cellules produit des quantités extraordinaires de données.
Une solution consiste à trier les cellules en fonction de leur teneur en ARN. "Les données unicellulaires sont composées d'un mélange sauvage de nombreux types de cellules différents. Nous nous intéressons aux cellules du même type cellulaire, qui devraient toutes se comporter de la même manière", explique Martin Vingron. Par conséquent, il est logique de regrouper des cellules similaires par calcul, dit-il. "Pour nous, les gènes marqueurs définissent un type de cellule."
Exploration interactive des amas de cellules
À l'aide de données accessibles au public sur les globules blancs, l'équipe a démontré le fonctionnement du nouvel algorithme. Les nombreux types différents de globules blancs comme les lymphocytes T, les lymphocytes B ou les monocytes sont tous regroupés en grappes distinctes. Les chercheurs ont confirmé des gènes marqueurs connus et ont pu montrer que des parents proches parmi les cellules sanguines partagent également une grande similitude dans leur activité génique.
"Chacun des gènes marqueurs que nous avons trouvés avec l'APL aurait pu être découvert par au moins une autre méthode existante pour l'identification des gènes marqueurs", explique Gralinska. Mais l'avantage d'APL par rapport aux algorithmes existants est sa représentation graphique des résultats, dit-elle. "Les outils existants fournissent de longues listes de gènes et de valeurs de score. Souvent, les utilisateurs parcourent la liste et s'arrêtent à un seuil arbitraire."
En revanche, la nouvelle méthode offre un moyen de visualiser ces gènes, de cliquer sur chacun d'eux et d'examiner de plus près son activité, dit-elle. "Nous ne fournissons pas seulement des listes de gènes marqueurs, nous permettons aux utilisateurs d'examiner le comportement de ces gènes", explique le chercheur. "Avec les diagrammes d'association, ils peuvent plonger dans leurs données pour en savoir plus sur chaque type de cellule." De plus, dit-elle, il est très facile de décomposer le rôle biologique des gènes les plus intéressants dans une étape ultérieure via l'analyse d'enrichissement des termes de Gene Ontology, qui est compatible avec le logiciel APL, ce qu'elle considère comme "une fonctionnalité très utile".
Le modèle mathématique sous-jacent
Les données de grande dimension qui contiennent des informations sur l'activité des gènes ne peuvent pas être représentées visuellement sans perte d'informations. Il en va de même pour les données groupées, ce qui complique l'analyse. "Notre astuce est que nous prenons en compte bien plus que deux ou trois dimensions, mais que nous créons finalement un diagramme en deux dimensions", explique Gralinska.
Les parcelles d'association sont dérivées d'une technique mathématique qui intègre simultanément les gènes et les cellules dans un espace commun de grande dimension. La mesure des distances entre les gènes et un groupe de cellules donné dans cet espace donne des paires de valeurs qui reflètent l'association d'un gène à un groupe donné et donnent un aperçu de son association à d'autres groupes.
"L'un des défauts de l'APL est que nous nous appuyons sur des données pré-groupées, ce qui signifie que nous devons nous appuyer sur d'autres techniques de clustering", explique Martin Vingron. "Néanmoins, nous espérons que notre nouvelle méthode trouvera de nombreux nouveaux utilisateurs. Nous constatons qu'un processus visuel et interactif permet tout simplement une meilleure analyse."