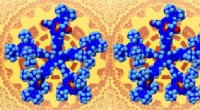
Instantané de la simulation MD de graphène sur Cu liquide. Crédit :Santiago Cingolani
Le graphène fait peut-être partie des découvertes scientifiques les plus passionnantes du siècle dernier. Bien qu'il nous soit étonnamment familier, le graphène est considéré comme un allotrope de carbone, ce qui signifie qu'il s'agit essentiellement de la même substance que le graphite mais dans une structure atomique différente - le graphène a également ouvert un nouveau monde de possibilités pour la conception et la construction de nouvelles technologies.
Le matériau est bidimensionnel, ce qui signifie que chaque "feuille" de graphène n'a que 1 atome d'épaisseur, mais ses liaisons le rendent aussi solide que certains des alliages métalliques les plus durs au monde tout en restant léger et flexible. Ce précieux, mélange unique de propriétés ont suscité l'intérêt des scientifiques d'un large éventail de domaines, menant à des recherches sur l'utilisation du graphène pour l'électronique de nouvelle génération, nouveaux revêtements sur instruments et outillages industriels, et les nouvelles technologies biomédicales.
C'est peut-être l'immense potentiel du graphène qui a par conséquent causé l'un de ses plus grands défis - le graphène est difficile à produire en gros volumes, et la demande pour le matériau ne cesse de croître. Des recherches récentes indiquent que l'utilisation d'un catalyseur au cuivre liquide peut être un moyen rapide, moyen efficace pour produire du graphène, mais les chercheurs n'ont qu'une compréhension limitée des interactions moléculaires qui se produisent au cours de ces brèves, moments chaotiques qui conduisent à la formation de graphène, ce qui signifie qu'ils ne peuvent pas encore utiliser la méthode pour produire de manière fiable des feuilles de graphène impeccables.
Afin de relever ces défis et d'aider à développer des méthodes pour une production de graphène plus rapide, une équipe de chercheurs de l'Université technique de Munich (TUM) a utilisé les systèmes de calcul haute performance (HPC) JUWELS et SuperMUC-NG au Jülich Supercomputing Center (JSC) et Leibniz Supercomputing Center (LRZ) pour exécuter une haute résolution simulations de formation de graphène sur cuivre liquide.
Une fenêtre sur l'expérimentation
L'attrait du graphène provient principalement de la structure cristalline parfaitement uniforme du matériau, ce qui signifie que produire du graphène avec des impuretés est un effort inutile. Pour les paramètres de laboratoire ou les circonstances où seule une petite quantité de graphène est nécessaire, les chercheurs peuvent placer un morceau de scotch sur un cristal de graphite et "décoller" les couches atomiques du graphite en utilisant une technique qui ressemble à la façon dont on utiliserait du ruban adhésif ou un autre adhésif pour aider à éliminer les poils d'animaux des vêtements. Bien que cela produise de manière fiable des couches de graphène impeccables, le processus est lent et peu pratique pour créer du graphène pour des applications à grande échelle.
L'industrie a besoin de méthodes qui pourraient produire de manière fiable du graphène de haute qualité moins cher et plus rapidement. L'une des méthodes les plus prometteuses à l'étude consiste à utiliser un catalyseur de métal liquide pour faciliter l'auto-assemblage d'atomes de carbone à partir de précurseurs moléculaires en une seule feuille de graphène poussant au-dessus du métal liquide. Alors que le liquide offre la possibilité d'augmenter efficacement la production de graphène, il introduit également une foule de complications, telles que les températures élevées requises pour faire fondre les métaux typiques utilisés, comme le cuivre.
Lors de la conception de nouveaux matériaux, les chercheurs utilisent des expériences pour voir comment les atomes interagissent dans diverses conditions. Alors que les progrès technologiques ont ouvert de nouvelles voies pour mieux comprendre le comportement à l'échelle atomique, même dans des conditions extrêmes telles que des températures très élevées, les techniques expérimentales ne permettent pas toujours aux chercheurs d'observer les réactions ultra-rapides qui facilitent les changements corrects de la structure atomique d'un matériau (ou quels aspects de la réaction peuvent avoir introduit des impuretés). C'est là que les simulations informatiques peuvent être utiles, cependant, simuler le comportement d'un système dynamique tel qu'un liquide n'est pas sans son lot de complications.
"Le problème pour décrire quelque chose comme ça est que vous devez appliquer des simulations de dynamique moléculaire (MD) pour obtenir le bon échantillonnage, " Andersen a dit. " Alors, bien sûr, il y a la taille du système - vous devez avoir un système suffisamment grand pour simuler avec précision le comportement du liquide." Contrairement aux expériences, les simulations de dynamique moléculaire offrent aux chercheurs la possibilité d'observer les événements qui se produisent à l'échelle atomique sous différents angles ou de mettre la simulation en pause pour se concentrer sur différents aspects.
Alors que les simulations MD offrent aux chercheurs un aperçu du mouvement des atomes individuels et des réactions chimiques qui n'ont pas pu être observées lors des expériences, ils ont leurs propres défis. Le principal d'entre eux est le compromis entre précision et coût - lorsqu'on s'appuie sur des méthodes ab initio précises pour piloter les simulations MD, il est extrêmement coûteux en termes de calcul d'obtenir des simulations suffisamment grandes et durent suffisamment longtemps pour modéliser avec précision ces réactions de manière significative.
Andersen et ses collègues ont utilisé environ 2, 500 cœurs sur JUWELS sur des périodes s'étalant sur plus d'un mois pour les simulations récentes. Malgré l'énorme effort de calcul, l'équipe ne pouvait encore simuler qu'environ 1, 500 atomes sur des picosecondes de temps. Bien que ces chiffres puissent sembler modestes, ces simulations étaient parmi les plus importantes des simulations ab initio MD de graphène sur cuivre liquide. L'équipe utilise ces simulations très précises pour aider à développer des méthodes moins coûteuses pour piloter les simulations MD afin qu'il devienne possible de simuler des systèmes plus grands et des échelles de temps plus longues sans compromettre la précision.
Renforcement des maillons de la chaîne
L'équipe a publié son travail de simulation record dans le Journal de physique chimique , ont ensuite utilisé ces simulations pour comparer avec les données expérimentales obtenues dans leur article le plus récent, qui est apparu dans ACS Nano .
Andersen a indiqué que les supercalculateurs de la génération actuelle, tels que JUWELS et SuperMUC-NG, permis à l'équipe d'effectuer sa simulation. Machines de nouvelle génération, cependant, ouvrirait encore plus de possibilités, car les chercheurs pourraient simuler plus rapidement de plus grands nombres ou systèmes sur de plus longues périodes.
Andersen a obtenu son doctorat. en 2014, et a indiqué que la recherche sur le graphène a explosé au cours de la même période. « Il est fascinant que le matériel soit un sujet de recherche si récent - il est presque encapsulé dans ma propre carrière scientifique que les gens l'aient examiné de près, ", a-t-elle déclaré. Malgré la nécessité de poursuivre les recherches sur l'utilisation de catalyseurs liquides pour produire du graphène, Andersen a indiqué que l'approche à deux volets consistant à utiliser à la fois le HPC et l'expérimentation serait essentielle pour poursuivre le développement du graphène et, à son tour, utilisation dans des applications commerciales et industrielles. « Dans cette recherche, il y a une grande interaction entre la théorie et l'expérience, et j'ai été des deux côtés de cette recherche, " elle a dit.