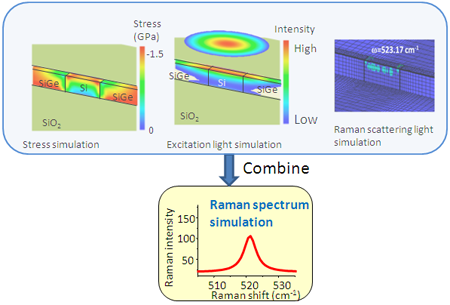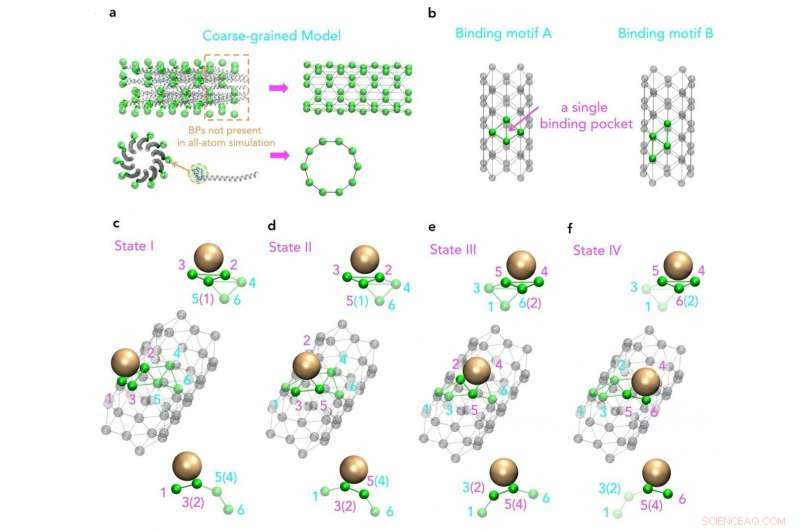
Vue moléculaire d'un modèle à gros grains basé sur la structure originale des protéines d'enveloppe majeures M13 Crédit :SUTD
Les simulations atomistiques sont un outil puissant pour étudier le mouvement et les interactions des atomes et des molécules. Dans de nombreux processus biologiques, effets à grande échelle, par exemple, l'assemblage de gros virus en nanoparticules est important. Les processus d'assemblage de ces grands virus sont d'une importance fondamentale pour la conception de nombreux dispositifs et thérapies ciblant les protéines virales. Cependant, l'échelle de temps et de longueur de ces processus d'assemblage est généralement trop grande pour des simulations à résolution moléculaire.
De plus, même si une augmentation de la puissance de calcul permet des simulations plus complexes et plus longues, structures virales, tels que M13, sont encore hors de portée. C'est pourquoi un groupe de recherche de l'Université de technologie et de design de Singapour (SUTD) et du Massachusetts Institute of Technology (MIT) a développé une procédure qui relie les processus d'assemblage à grande échelle aux simulations moléculaires. Professeur adjoint Desmond Loke de la Science de SUTD, Le cluster Mathématiques et Technologie a déclaré :"Pour la simulation de M13, nous avons commencé avec différents ensembles de champs de force. Des champs de force appropriés ont été choisis et ils ont été utilisés comme entrées pour des simulations de dynamique de molécule avec le modèle à gros grains conçu pour capturer le modèle clé du processus d'assemblage. »
« Bien que nous sachions que la fabrication à base de M13 peut être fondamentalement entraînée par des interactions nanoparticule-peptide, qui peut aussi être un principe clé de la bio-ingénierie de type M13, nous avons peu de connaissances sur la façon dont les motifs répétés de peptides à extrémité courte sur une surface M13 sont réellement impliqués dans ces interactions. Pour étudier cela, nous devons idéalement inclure une structure complète de la protéine d'enveloppe virale, ce qui est une tâche difficile pour les simulations actuelles de dynamique moléculaire de pointe, " ajoute le Dr Luna Li, premier auteur de l'article.
La procédure permet aux utilisateurs d'ajouter différents types de nanoparticules à une solution, à un niveau réaliste. Inspiré par cette procédure, Le professeur adjoint Loke et ses collègues ont pu simuler un virus à grande échelle avec des nanoparticules et à l'intérieur d'une solution pendant cinquante nanosecondes.
Le Dr Li a dit :"La structure et la solution du virus en contiennent environ 700, 000 atomes au total." Compte tenu de la forme et de la taille des caractéristiques, la complexité de cette simulation peut être plus grande que n'importe quelle simulation effectuée précédemment.
"Une simulation réalisée en microsecondes aurait été possible si un modèle M13 plus petit avait été utilisé, mais il peut être intéressant de réduire le temps pour observer réellement comment la structure complète peut influencer l'assemblage entre le M13 et les nanoparticules, " a expliqué le professeur adjoint Loke.