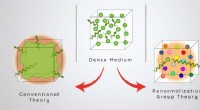
Les matériaux sont composés d’atomes composés de protons, de neutrons et d’électrons. Les interactions entre ces particules déterminent les propriétés du matériau, telles que sa résistance, sa conductivité et son comportement magnétique. Comprendre ces interactions est essentiel pour concevoir de nouveaux matériaux dotés des propriétés souhaitées pour un large éventail d'applications, telles que le stockage d'énergie, l'électronique et la catalyse.
L’une des méthodes les plus précises pour étudier le comportement des électrons dans les matériaux est la théorie fonctionnelle de la densité (TFD), qui est une méthode largement utilisée pour calculer la structure électronique des atomes, des molécules et des solides. Cependant, les calculs DFT peuvent nécessiter beaucoup de calculs, en particulier pour les grands systèmes ou ceux contenant des éléments lourds, ce qui les rend difficiles à appliquer dans de nombreux cas pratiques.
L'approche du champ auto-cohérent (SCF) implique la résolution des équations de Kohn-Sham, un ensemble d'équations qui définissent les calculs DFT. Dans l'approche traditionnelle, les équations de Kohn-Sham sont résolues en développant les fonctions d'onde des électrons dans un ensemble fini de fonctions de base, telles que les ondes planes. Cette approche peut être coûteuse en termes de calcul, en particulier pour les systèmes comportant un grand nombre d’atomes.
La nouvelle technique développée par les chercheurs d'Argonne utilise une approche plus efficace appelée jeu de base des ondes planes. Dans cette approche, les fonctions d'onde sont représentées sur une grille puis projetées sur un ensemble d'ondes planes. Cela réduit le coût des calculs et permet aux scientifiques d’étudier des systèmes plus vastes avec une plus grande précision et efficacité.
"Le développement de cette nouvelle technique constitue une avancée majeure dans le domaine de la science informatique des matériaux", a déclaré le Dr John Perdew, scientifique principal à Argonne et l'un des principaux chercheurs de l'étude. "Cela ouvre la porte à de nouvelles possibilités pour étudier le comportement des électrons dans les matériaux, ce qui accélérera le développement de matériaux avancés."
Les chercheurs ont démontré la puissance de leur nouvelle technique en étudiant divers matériaux, notamment le silicium, l’eau et un oxyde complexe. Ils ont constaté que leur technique peut atteindre une précision similaire aux calculs DFT traditionnels, mais avec un coût de calcul considérablement réduit, ce qui en fait un outil prometteur pour la recherche future sur les matériaux.
L'étude, intitulée « Théorie fonctionnelle de densité de champ auto-cohérente avec un ensemble de bases d'ondes planes :formalisme et mise en œuvre », a été publiée dans le Journal of Chemical Physics et a été soutenue par le Bureau des sciences du DOE. L'équipe de recherche comprenait des scientifiques du Laboratoire national d'Argonne, de l'Université de Californie à Berkeley et de l'Université de l'Illinois à Urbana-Champaign.