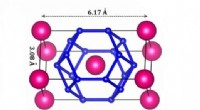
La distribution électronique tétraédrique d'une molécule d'eau. Le noyau de l'atome d'oxygène est au centre du tétraèdre, et les noyaux d'hydrogène sont au centre des sphères roses. Fondation Simons. Crédit :Fondation Simons
Un nouvel outil d'apprentissage automatique peut calculer l'énergie nécessaire pour fabriquer ou casser une molécule avec une plus grande précision que les méthodes conventionnelles. Alors que l'outil ne peut actuellement traiter que des molécules simples, il ouvre la voie à de futures perspectives en chimie quantique.
"Utiliser l'apprentissage automatique pour résoudre les équations fondamentales régissant la chimie quantique est un problème ouvert depuis plusieurs années, et il y a beaucoup d'excitation autour de ça en ce moment, " dit le co-créateur Giuseppe Carleo, chercheur au Centre de physique quantique computationnelle du Flatiron Institute à New York. Une meilleure compréhension de la formation et de la destruction des molécules, il dit, pourrait révéler le fonctionnement interne des réactions chimiques essentielles à la vie.
Carleo et ses collaborateurs Kenny Choo de l'Université de Zurich et Antonio Mezzacapo du IBM Thomas J. Watson Research Center à Yorktown Heights, New York, présentent leur travail le 12 mai à Communication Nature .
L'outil de l'équipe estime la quantité d'énergie nécessaire pour assembler ou séparer une molécule, comme l'eau ou l'ammoniaque. Ce calcul nécessite de déterminer la structure électronique de la molécule, qui consiste en le comportement collectif des électrons qui lient la molécule entre eux.
La structure électronique d'une molécule est une chose délicate à calculer, nécessitant la détermination de tous les états potentiels dans lesquels les électrons de la molécule pourraient se trouver, plus la probabilité de chaque état.
Puisque les électrons interagissent et s'entremêlent mécaniquement quantiquement, les scientifiques ne peuvent pas les traiter individuellement. Avec plus d'électrons, plus d'enchevêtrements surgissent, et le problème devient exponentiellement plus difficile. Il n'existe pas de solutions exactes pour des molécules plus complexes que les deux électrons trouvés dans une paire d'atomes d'hydrogène. Même les approximations ont du mal à être précises lorsqu'elles impliquent plus que quelques électrons.
L'un des défis est que la structure électronique d'une molécule comprend des états pour un nombre infini d'orbitales allant de plus en plus loin des atomes. En outre, un électron est indiscernable d'un autre, et deux électrons ne peuvent pas occuper le même état. Cette dernière règle est une conséquence de la symétrie d'échange, qui régit ce qui se passe lorsque des particules identiques changent d'état.
Mezzacapo et ses collègues d'IBM Quantum ont développé une méthode pour contraindre le nombre d'orbitales considérées et imposer une symétrie d'échange. Cette approche, basé sur des méthodes développées pour des applications d'informatique quantique, rend le problème plus proche des scénarios où les électrons sont confinés à des emplacements prédéfinis, comme dans un treillis rigide.
La similitude avec les treillis rigides était la clé pour rendre le problème plus gérable. Carleo a précédemment formé des réseaux de neurones pour reconstruire le comportement des électrons confinés aux sites d'un réseau. En étendant ces méthodes, les chercheurs ont pu estimer des solutions aux problèmes compactés de Mezzacapo. Le réseau de neurones de l'équipe calcule la probabilité de chaque état. En utilisant cette probabilité, les chercheurs peuvent estimer l'énergie d'un état donné. Le niveau d'énergie le plus bas, appelé l'énergie d'équilibre, C'est là que la molécule est la plus stable.
Les innovations de l'équipe ont rendu le calcul de la structure électronique d'une molécule de base plus simple et plus rapide. Les chercheurs ont démontré la précision de leurs méthodes en estimant la quantité d'énergie qu'il faudrait pour séparer une molécule du monde réel, rompre ses liens. Ils ont effectué des calculs pour le dihydrogène (H
À l'avenir, les chercheurs visent à s'attaquer à des molécules plus grosses et plus complexes en utilisant des réseaux de neurones plus sophistiqués. L'un des objectifs est de gérer des produits chimiques comme ceux que l'on trouve dans le cycle de l'azote, dans lequel les processus biologiques construisent et brisent des molécules à base d'azote pour les rendre utilisables pour la vie. "Nous voulons que ce soit un outil qui puisse être utilisé par les chimistes pour traiter ces problèmes, " dit Carléo.
Carléo, Choo et Mezzacapo ne sont pas les seuls à exploiter l'apprentissage automatique pour résoudre les problèmes de la chimie quantique. Les chercheurs ont présenté leurs travaux pour la première fois sur arXiv.org en septembre 2019. Ce même mois, un groupe en Allemagne et un autre à DeepMind de Google à Londres ont chacun publié des recherches utilisant l'apprentissage automatique pour reconstruire la structure électronique des molécules.
Les deux autres groupes utilisent une approche similaire qui ne limite pas le nombre d'orbitales considérées. Cette inclusion, cependant, est plus exigeant en termes de calcul, un inconvénient qui ne fera que s'aggraver avec des molécules plus complexes. Avec les mêmes ressources de calcul, l'approche de Carleo, Choo et Mezzacapo offrent une plus grande précision, mais les simplifications apportées pour obtenir cette précision pourraient introduire des biais.
"Globalement, c'est un compromis entre biais et précision, et on ne sait pas laquelle des deux approches a le plus de potentiel pour l'avenir, " dit Carleo. " Seul le temps nous dira laquelle de ces approches peut être adaptée aux problèmes ouverts difficiles de la chimie. "