
Shantenu Jha, président du Brookhaven Lab's Center for Data-Driven Discovery, et son équipe de l'Université Rutgers et de l'University College London ont conçu un cadre logiciel pour calculer avec précision et rapidité la force avec laquelle les candidats-médicaments se lient à leurs protéines cibles. Le cadre vise à résoudre le problème réel de la conception de médicaments - actuellement un processus long et coûteux - et pourrait avoir un impact sur la médecine personnalisée. Crédit :Laboratoire national de Brookhaven
Les solutions à de nombreux problèmes scientifiques et techniques du monde réel, de l'amélioration des modèles météorologiques à la conception de nouveaux matériaux énergétiques en passant par la compréhension de la formation de l'univers, nécessitent des applications pouvant atteindre une très grande taille et des performances élevées. Chaque année, à travers son International Scalable Computing Challenge (SCALE), l'Institut des ingénieurs électriciens et électroniciens (IEEE) reconnaît un projet qui fait progresser le développement d'applications et l'infrastructure de soutien pour permettre le développement à grande échelle, calcul haute performance nécessaire pour résoudre de tels problèmes.
Le gagnant de cette année, « Permettre un compromis entre la précision et le coût de calcul :des algorithmes adaptatifs pour réduire le délai d'obtention d'un aperçu clinique, " est le résultat d'une collaboration entre des chimistes et des informaticiens et informaticiens du laboratoire national de Brookhaven du Département de l'énergie des États-Unis (DOE), Université Rutgers, et University College de Londres. Les membres de l'équipe ont été honorés lors du 18e IEEE/Association for Computing Machinery (ACM) International Symposium on Cluster, Cloud et Grid Computing à Washington, DC, du 1er au 4 mai.
« Nous avons développé une méthodologie de calcul numérique pour évaluer précisément et rapidement l'efficacité de différents candidats médicaments, " a déclaré Shantenu Jha, membre de l'équipe, président du Center for Data-Driven Discovery, partie de l'initiative de science informatique de Brookhaven Lab. "Bien que nous n'ayons pas encore appliqué cette méthodologie pour concevoir un nouveau médicament, nous avons démontré qu'il pouvait fonctionner aux grandes échelles impliquées dans le processus de découverte de médicaments."
La découverte de médicaments, c'est un peu comme concevoir une clé pour s'adapter à une serrure. Pour qu'un médicament soit efficace dans le traitement d'une maladie particulière, il doit se lier étroitement à une molécule, généralement une protéine, associée à cette maladie. Ce n'est qu'alors que le médicament peut activer ou inhiber la fonction de la molécule cible. Les chercheurs peuvent en filtrer 10, 000 ou plus de composés moléculaires avant de trouver ceux qui ont l'activité biologique souhaitée. Mais ces composés "plombants" manquent souvent de puissance, sélectivité, ou la stabilité nécessaire pour devenir un médicament. En modifiant la structure chimique de ces plombs, les chercheurs peuvent concevoir des composés dotés des propriétés médicamenteuses appropriées. Les candidats-médicaments conçus passent ensuite le long du pipeline de développement jusqu'à l'étape des tests précliniques. Parmi ces candidats, seule une petite fraction entre dans la phase d'essai clinique, et un seul finit par devenir un médicament approuvé à l'usage des patients. La mise sur le marché d'un nouveau médicament peut prendre une décennie ou plus et coûter des milliards de dollars.
Surmonter les goulots d'étranglement de la conception de médicaments grâce à la science informatique
Les progrès récents de la technologie et des connaissances ont ouvert une nouvelle ère de découverte de médicaments, une ère qui pourrait réduire considérablement le temps et les dépenses du processus de développement de médicaments. L'amélioration de notre compréhension des structures cristallines 3-D des molécules biologiques et l'augmentation de la puissance de calcul permettent d'utiliser des méthodes informatiques pour prédire les interactions médicament-cible.
La découverte de médicaments est un problème de verrouillage et de clé dans lequel le médicament (clé) doit s'adapter spécifiquement à la cible biologique (verrou). Crédit :Laboratoire national de Brookhaven
En particulier, une technique de simulation informatique appelée dynamique moléculaire s'est révélée prometteuse pour prédire avec précision la force avec laquelle les molécules médicamenteuses se lient à leurs cibles (affinité de liaison). La dynamique moléculaire simule la façon dont les atomes et les molécules se déplacent lorsqu'ils interagissent dans leur environnement. En cas de découverte de médicaments, les simulations révèlent comment les molécules médicamenteuses interagissent avec leur protéine cible et modifient la conformation de la protéine, ou forme, qui détermine sa fonction.
Cependant, ces capacités de prédiction ne fonctionnent pas encore à une échelle suffisamment grande ou à une vitesse suffisamment rapide pour que les sociétés pharmaceutiques les adoptent dans leur processus de développement.
« Traduire ces avancées en matière de précision prédictive pour avoir un impact sur la prise de décision industrielle nécessite que, de l'ordre de 10, 000 affinités de liaison sont calculées au plus vite, sans perte de précision, " a déclaré Jha. " Produire des informations en temps opportun exige une efficacité de calcul qui repose sur le développement de nouveaux algorithmes et de systèmes logiciels évolutifs, et l'allocation intelligente des ressources de calcul intensif."
Jha et ses collaborateurs de l'Université Rutgers, où il est également professeur au Département de génie électrique et informatique, et University College London ont conçu un cadre logiciel pour prendre en charge le calcul précis et rapide des affinités de liaison tout en optimisant l'utilisation des ressources de calcul. Ce cadre, appelé High-Throughput Binding Affinity Calculator (HTBAC), s'appuie sur le projet RADICAL-Cybertools que Jha dirige en tant que chercheur principal de Rutgers' Research in Advanced Distributed Cyberinfrastructure and Applications Laboratory (RADICAL). L'objectif de RADICAL-Cybertools est de fournir une suite de briques logicielles pour prendre en charge les workflows d'applications scientifiques à grande échelle sur des plateformes de calcul haute performance, qui agrègent la puissance de calcul pour résoudre de gros problèmes de calcul qui seraient autrement insolubles en raison du temps requis.
En informatique, les workflows font référence à une série d'étapes de traitement nécessaires pour accomplir une tâche ou résoudre un problème. Surtout pour les flux de travail scientifiques, il est important que les workflows soient flexibles afin qu'ils puissent s'adapter dynamiquement pendant l'exécution pour fournir les résultats les plus précis tout en utilisant efficacement le temps de calcul disponible. De tels flux de travail adaptatifs sont idéaux pour la découverte de médicaments, car seuls les médicaments ayant des affinités de liaison élevées doivent être évalués plus avant.
« Le compromis souhaité entre la précision requise et le coût (temps) de calcul change tout au long de la découverte du médicament au fur et à mesure que le processus passe du dépistage à la sélection des pistes, puis à l'optimisation des pistes, " a déclaré Jha. " Un nombre important de composés doivent être sélectionnés à moindre coût pour éliminer les mauvais liants avant que des méthodes plus précises ne soient nécessaires pour distinguer les meilleurs liants. Fournir le délai de résolution le plus rapide nécessite de surveiller l'avancement des simulations et de fonder les décisions concernant la poursuite de l'exécution sur l'importance scientifique."
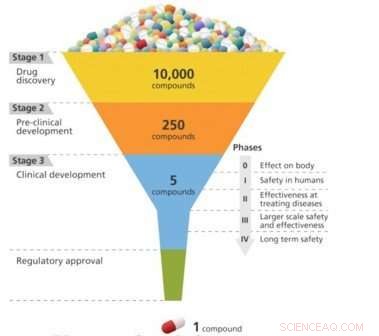
Un schéma du processus de développement d'un médicament, qui se concentre progressivement sur les candidats les plus efficaces d'un large bassin initial. Crédit :Laboratoire national de Brookhaven
En d'autres termes, il n'aurait pas de sens de continuer les simulations d'une interaction médicament-protéine particulière si le médicament se lie faiblement à la protéine par rapport aux autres candidats. Mais il serait logique d'allouer des ressources de calcul supplémentaires si un médicament présente une affinité de liaison élevée.
La prise en charge des flux de travail adaptatifs à grande échelle caractéristiques des programmes de découverte de médicaments nécessite des capacités de calcul avancées. HTBAC fournit une telle prise en charge via une couche logicielle middleware flexible qui permet l'exécution adaptative des algorithmes. Actuellement, HTBAC prend en charge deux algorithmes :échantillonnage amélioré de la dynamique moléculaire avec approximation du solvant continu (ESMACS) et intégration thermodynamique avec échantillonnage amélioré (TIES). ESMACS, une méthode de calcul moins coûteuse mais moins rigoureuse que TIES, calcule la force de liaison d'un médicament à sa protéine cible sur la base de simulations de dynamique moléculaire. Par contre, TIES compare les affinités de liaison relatives de deux médicaments différents à la même protéine.
"ESMACS fournit une approche quantitative rapide suffisamment sensible pour déterminer les affinités de liaison afin que nous puissions éliminer les mauvais liants, tandis que TIES fournit une méthode plus précise pour étudier les bons liants à mesure qu'ils sont raffinés et améliorés, " dit Jumana Dakka, un doctorat de deuxième année. étudiant à Rutgers et membre du groupe RADICAL.
Afin de déterminer quel algorithme exécuter, HTBAC analyse les calculs d'affinité de liaison au moment de l'exécution. Cette analyse éclaire les décisions concernant le nombre de simulations simultanées à effectuer et si des étapes de stimulation doivent être ajoutées ou supprimées pour chaque candidat médicament étudié.
Mettre le cadre à l'épreuve
Jha's team demonstrated how HTBAC could provide insight from drug candidate data on a short timescale by reproducing results from a collaborative study between University College London and the London-based pharmaceutical company GlaxoSmithKline to discover drug compounds that bind to the BRD4 protein. Known to play a key role in driving cancer and inflammatory diseases, the BRD4 protein is a major target of bromodomain-containing (BRD) inhibitors, a class of pharmaceutical drugs currently being evaluated in clinical trials. The researchers involved in this collaborative study are focusing on identifying promising new drugs to treat breast cancer while developing an understanding of why certain drugs fail in the presence of breast cancer gene mutations.
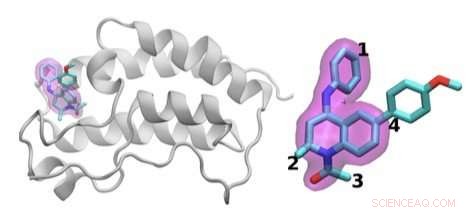
The scientists investigated the chemical structures of 16 drugs based on the same tetrahydroquinoline (THQ) scaffold. On the left is a cartoon of the BRD4 protein bound to one of these drugs; on the right is a molecular representation of a drug with the THQ scaffold highlighted in magenta. Regions that are chemically modified between the drugs investigated in this study are labeled 1 to 4. Typically, only a small change is made to the chemical structure of one drug to the next. This conservative approach makes it easier for researchers to understand why one drug is effective, whereas another is not. Crédit :Laboratoire national de Brookhaven
Jha and his team concurrently screened a group of 16 closely related drug candidates from the study by running thousands of computational sequences on more than 32, 000 computing cores. They ran the computations on the Blue Waters supercomputer at the National Center for Computing Applications, University of Illinois at Urbana-Champaign.
In a real drug design scenario, many more compounds with a wider range of chemical properties would need to be investigated. The team members previously demonstrated that the workload management layer and runtime system underlying HTBAC could scale to handle 10, 000 concurrent tasks.
"HTBAC could support the concurrent screening of different compounds at unprecedented scales—both in the number of compounds and computational resources used, " said Jha. "We showed that HTBAC has the ability to solve a large number of drug candidates in essentially the same amount of time it would take to solve a smaller set, assuming the number of processors increases proportionally with the number of candidates."
This ability is made possible through HTBAC's adaptive functionality, which allows it to execute the optimal algorithm depending on the properties of the drugs being investigated, improving the accuracy of the results and minimizing compute time.
"The lead optimization stage usually considers on the order of 10, 000 small molecules, " said Jha. "While experiment automation reduces the amount of time needed to calculate the binding affinities, HTBAC has the potential to cut this time (and cost) by an order of magnitude or more."
With HTBAC, TIES requires approximately 25, 000 central processing unit (CPU) core hours for a single prediction. At least a 250 million core hours would be needed for a large-scale study to support a pharmaceutical drug screening campaign, with a typical turnaround time of about two weeks. HTBAC could facilitate running studies requiring sustained usage of millions of core hours per day.
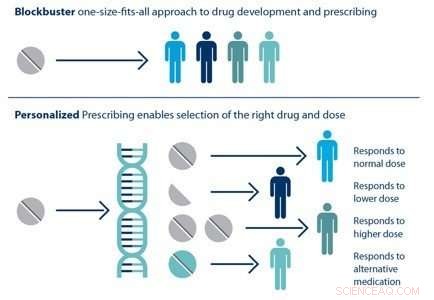
Individual patients respond differently to drugs. The ability to predict which treatment is best for a particular patient based on his or her genetic sequence is the goal of personalized medicine. Crédit :Laboratoire national de Brookhaven
When the University of College London–GlaxoSmithKline study concludes, Jha and his team hope to be given the experimental data on the tens of thousands of drug candidates, without knowing which candidate ended up being the best one. Avec ces informations, they could perform a blind test to determine whether HTBAC provides an improvement in compute time (for a given accuracy) over the existing automated methods for drug discovery. If necessary, they could then refine their methodology.
Applying scalable computing to precision medicine
HTBAC not only has the potential to improve the speed and accuracy of drug discovery in the pharmaceutical industry but also to improve individual patient outcomes in clinical settings. Using target proteins based on a patient's genetic sequence, HTBAC could predict a patient's response to different drug treatments. This personalized assessment could replace the traditional one-size-fits-all approach to medicine. Par exemple, such predictions could help determine which cancer patients would actually benefit from chemotherapy, avoiding unnecessary toxicity.
According to Jha, the computation time would have to be significantly reduced in order for physicians to clinically use HTBAC to treat their patients:"Our grand vision is to apply scalable computing techniques to personalized medicine. If we can use these techniques to optimize drugs and drug cocktails for each individual's unique genetic makeup on the order of a few days, we will be empowered to treat diseases much more effectively."
"Extreme-scale computing for precision medicine is an emerging area that CSI and Brookhaven at large have begun to tackle, " said CSI Director Kerstin Kleese van Dam. "This work is a great example of how technologies we originally developed to tackle DOE challenges can be applied to other domains of high national impact. We look forward to forming more strategic partnerships with other universities, pharmaceutical companies, and medical institutions in this important area that will transform the future of health care."














