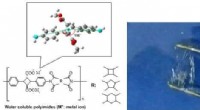
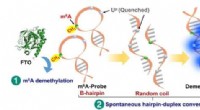
L’équipe de recherche a utilisé des calculs de théorie fonctionnelle de la densité (DFT) pour étudier le comportement des molécules d’eau en présence d’ions. DFT est un outil puissant pour simuler la structure électronique des matériaux et des molécules. L’équipe s’est concentrée sur deux systèmes spécifiques :la solvatation des ions lithium (Li+) et l’interaction des molécules d’eau avec une surface lithium-oxygène (Li-O).
Dans le cas de la solvatation Li+, les calculs DFT ont révélé que les molécules d’eau forment une coque d’hydratation hautement ordonnée autour de l’ion lithium. Cette structure ordonnée résulte des fortes interactions électrostatiques entre l’ion lithium chargé positivement et les atomes d’oxygène chargés négativement des molécules d’eau. La coque d'hydratation empêche efficacement l'ion lithium d'interagir avec d'autres ions ou molécules dans la solution, ce qui est crucial pour stabiliser l'ion et faciliter son transport dans les réactions électrochimiques.
En outre, les chercheurs ont étudié l’interaction des molécules d’eau avec une surface Li-O, qui représente la surface des électrodes d’une batterie lithium-ion. Les calculs DFT ont montré que les molécules d’eau forment de fortes liaisons hydrogène avec les atomes d’oxygène à la surface, créant ainsi un réseau d’eau qui bloque efficacement la migration des ions lithium depuis l’électrode. Cet effet de blocage est responsable de la passivation de la surface de l'électrode et de la réduction des performances de la batterie au fil du temps.
Dans l’ensemble, le cadre théorique développé par l’équipe de recherche fournit une compréhension globale du rôle des molécules d’eau dans la synthèse des matériaux et dans les processus de fabrication impliquant des ions. Les résultats contribuent à la conception rationnelle et à l’optimisation des réactions électrochimiques et des systèmes de batteries, permettant ainsi d’obtenir des matériaux et des dispositifs plus efficaces et plus durables.