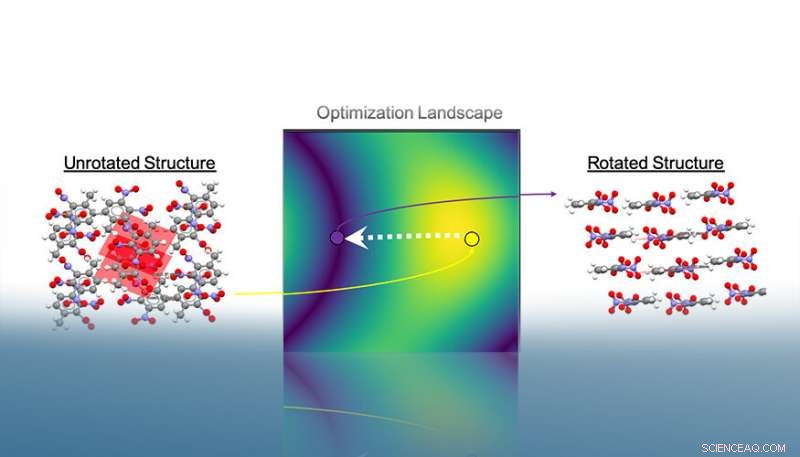
Autopack fait tourner les structures cristallines dans l'espace 3D pour minimiser la surface projetée de leurs molécules. Après convergence, il est possible d'extraire le motif de tassement associé au cristal en fonction des angles interplanaires relatifs. Dans cet exemple, les empilements trouvés après la procédure d'optimisation indiquent le motif d'emballage bêta de la structure. Crédit :Laboratoire national Lawrence Livermore
Que les chimistes organiques travaillent au développement d'une nouvelle énergie moléculaire ou à la création de nouveaux médicaments à succès dans l'industrie pharmaceutique, chacun cherche comment optimiser la structure chimique d'une molécule pour atteindre les propriétés cibles souhaitées.
Une partie de cette optimisation comprend le motif d'emballage d'un cristal moléculaire, un modèle perçu dans la façon dont les molécules s'orientent les unes par rapport aux autres dans une structure cristalline. Les ensembles de données actuels sur les motifs d'emballage sont restés petits en raison de processus d'étiquetage manuels intensifs et de schémas d'étiquetage insuffisants.
Pour aider à résoudre ce problème, une équipe de matériaux et d'informaticiens du Lawrence Livermore National Laboratory (LLNL) a développé un package disponible gratuitement, Emballage automatique, qui formalise le processus d'étiquetage des motifs d'emballage et peut automatiquement traiter et étiqueter les motifs d'emballage de milliers de structures cristallines moléculaires. La recherche apparaît dans le Journal of Chemical Information and Modelling .
Des études d'ingénierie cristalline à petite échelle au cours des 30 dernières années suggèrent que, alors que prédire des structures cristallines expérimentales à partir d'une seule structure chimique reste hors de portée, il peut y avoir des relations entre les structures chimiques des molécules et un attribut spécifique de la structure cristalline qu'elles adoptent appelé le motif d'emballage.
Le motif d'emballage d'un cristal moléculaire est un concept important pour les applications énergétiques et électroniques organiques en raison des corrélations observées entre les motifs d'emballage des cristaux moléculaires et les propriétés de performance d'intérêt, qui incluent l'insensibilité pour les explosifs moléculaires et le transport de charge pour les semi-conducteurs moléculaires.
Aucune méthode formalisée et open source d'attribution de motifs d'emballage n'a été créée jusqu'à présent. Au lieu, les motifs d'emballage sont attribués aux cristaux moléculaires simplement par l'évaluation humaine d'une structure cristalline et le jugement, résultant en des ensembles de données petits et bruyants.
« À l'ère de l'apprentissage automatique, la capacité de créer de grandes, les ensembles de données étiquetés de motifs d'emballage de cristaux moléculaires sont maintenant particulièrement importants, " a déclaré le data scientist du LLNL Donald Loveland, auteur principal de l'article. "De tels efforts peuvent générer des modèles qui peuvent prédire les motifs d'emballage à partir de la structure chimique des molécules uniquement, ce qui aiderait les chimistes organiques à prioriser les synthèses de nouvelles molécules en fonction du motif et des propriétés d'emballage souhaités."
Le nouveau travail LLNL utilise un algorithme d'optimisation efficace qui contourne de nombreux problèmes rencontrés dans les méthodes d'étiquetage des motifs d'emballage proposées précédemment, conduisant à de nouveaux résultats de pointe lorsqu'ils sont testés sur un ensemble de données organisé par LLNL.
Grâce à l'emballage automatique, les chercheurs ont pu générer un ensemble de données de près de 10, 000 motifs d'emballage pour un ensemble de molécules énergétiques et quasi-énergétiques d'intérêt pour le Lab, une tâche qui aurait été impossible auparavant. Pour le contexte, la littérature précédente est restée limitée à l'ordre de 100 molécules en raison de la nature fastidieuse et chronophage de l'étiquetage manuel. Les premières analyses de ce nouvel ensemble de données suggèrent des tendances complexes entre les interactions intermoléculaires, Des conformations moléculaires 3D et des motifs de garnissage adoptés actuellement inexplorés sur le terrain, fournir des conseils sur les prochaines étapes pour les pipelines d'ingénierie cristalline.
Le code est disponible gratuitement auprès du Bureau Innovations et Partenariats du Lab.