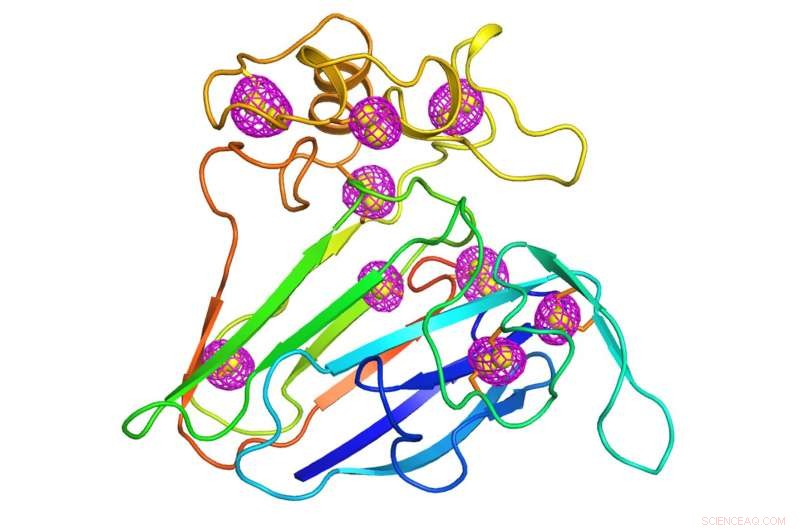
Un dessin animé représentant la structure d'une protéine végétale bien étudiée qui a servi de cas de test pour la technique de microcristallographie nouvellement développée. Les motifs de maille magenta entourant les atomes de soufre intrinsèques à la protéine (sphères jaunes) indiquent les signaux anormaux qui ont été extraits par diffraction des rayons X à basse énergie de milliers de cristaux mesurant moins de 10 millionièmes de mètre, la taille d'une bactérie. Crédit :Laboratoire national de Brookhaven
L'utilisation des rayons X pour révéler les structures 3D à l'échelle atomique des protéines a conduit à d'innombrables progrès dans la compréhension du fonctionnement de ces molécules dans les bactéries, virus, les plantes, et les humains - et a guidé le développement de médicaments de précision pour lutter contre des maladies telles que le cancer et le sida. Mais de nombreuses protéines ne peuvent pas être transformées en cristaux suffisamment gros pour que leurs arrangements atomiques soient déchiffrés. Pour relever ce défi, Des scientifiques du laboratoire national de Brookhaven du Département de l'énergie des États-Unis (DOE) et des collègues de l'Université Columbia ont développé une nouvelle approche pour résoudre les structures protéiques à partir de minuscules cristaux.
La méthode repose sur une manipulation unique des échantillons, extraction de signal, et les approches d'assemblage de données, et une ligne de faisceau capable de focaliser des rayons X intenses à Brookhaven's National Synchrotron Light Source II (NSLS-II) - une installation utilisateur du DOE Office of Science - à un point d'un millionième de mètre, environ un cinquantième de la largeur d'un cheveu humain.
"Notre technique ouvre vraiment la porte au traitement de microcristaux jusque-là inaccessibles, y compris les récepteurs de surface cellulaire difficiles à cristalliser et d'autres protéines membranaires, protéines souples, et de nombreuses protéines humaines complexes, " a déclaré Qun Liu, scientifique du Brookhaven Lab, l'auteur correspondant de l'étude, qui a été publié le 3 mai, 2019, dans IUCrJ , une revue de l'Union Internationale de Cristallographie.
Déchiffrer les structures des protéines
La cristallographie des protéines est une méthode dominante pour résoudre les structures des protéines depuis 1958, s'améliorant au fil du temps à mesure que les sources de rayons X sont devenues plus puissantes, permettant des déterminations de structure plus précises. Pour déterminer la structure d'une protéine, les scientifiques mesurent comment les rayons X comme ceux générés à NSLS-II diffractent, ou rebondir, les atomes d'un réseau cristallin ordonné constitué de nombreuses copies de la même molécule de protéine sont tous disposés de la même manière. Le diagramme de diffraction donne des informations sur l'emplacement des atomes. Mais ce n'est pas suffisant.
"Seules les amplitudes des 'ondes' de rayons X diffractées sont enregistrées sur le détecteur, mais pas leurs phases (le timing entre les vagues), " a déclaré Liu. " Les deux sont nécessaires pour reconstruire une structure en 3D. C'est ce qu'on appelle le problème de la phase cristallographique."
Les cristallographes ont résolu ce problème en collectant des données de phase à partir d'un autre type de diffusion, connu sous le nom de diffusion anormale. La diffusion anormale se produit lorsque des atomes plus lourds que les principaux composants de carbone d'une protéine, hydrogène, et l'azote absorbe et réémet une partie des rayons X. Cela se produit lorsque l'énergie des rayons X est proche de l'énergie que ces atomes lourds aiment absorber. Les scientifiques insèrent parfois artificiellement des atomes lourds tels que le sélénium ou le platine dans la protéine à cette fin. Mais les atomes de soufre, qui apparaissent naturellement dans les molécules de protéines, peut également produire de tels signaux, quoique plus faible. Même si ces signaux anormaux sont faibles, un gros cristal a généralement suffisamment de copies de la protéine avec suffisamment d'atomes de soufre pour les rendre mesurables. Cela donne aux scientifiques les informations de phase nécessaires pour localiser l'emplacement des atomes de soufre et traduire les schémas de diffraction en une structure 3-D complète.
"Une fois que vous connaissez les positions du soufre, vous pouvez calculer les phases pour les autres atomes de protéines car la relation entre le soufre et les autres atomes est fixe, " dit Liu.
Mais de minuscules cristaux, par définition, n'ont pas autant de copies de la protéine d'intérêt. Ainsi, au lieu de rechercher des informations de diffraction et de phase à partir de copies répétées d'une protéine dans un seul grand cristal, l'équipe de Brookhaven/Columbia a développé un moyen de prendre des mesures à partir de nombreux cristaux minuscules, puis assembler les données collectives.
Petits cristaux, gros résultats
Pour manipuler les minuscules cristaux, l'équipe a développé des grilles d'échantillons à motifs de puits de petite taille. Après avoir versé le solvant contenant les microcristaux sur ces grilles bien montées, les scientifiques ont retiré le solvant et ont gelé les cristaux qui étaient piégés sur les grilles.
"Nous avons encore un défi, bien que, parce que nous ne pouvons pas voir où se trouvent les minuscules cristaux sur notre grille, " dit Liu. " Pour le savoir, nous avons utilisé la microdiffraction sur la ligne de faisceau de la cristallographie macromoléculaire à microfocalisation frontalière (FMX) du NSLS-II pour étudier l'ensemble de la grille. Balayage ligne par ligne, nous pouvons trouver où ces cristaux sont cachés."
Comme Martin Fuchs, le scientifique principal de la ligne de lumière chez FMX, expliqué, "La ligne de lumière FMX peut focaliser toute l'intensité du faisceau de rayons X jusqu'à une taille d'un micron, ou millionième de mètre. Nous pouvons contrôler finement la taille du faisceau pour l'adapter à la taille des cristaux, cinq microns dans le cas de l'expérience actuelle. Ces capacités sont cruciales pour obtenir le meilleur signal, " il a dit.
Wuxian Shi, un autre scientifique de la ligne de lumière FMX, a noté que « les données recueillies dans l'enquête sur la grille contiennent des informations sur l'emplacement des cristaux. En outre, nous pouvons également voir à quel point chaque cristal diffracte, ce qui nous permet de ne sélectionner que les meilleurs cristaux pour la collecte de données."
Les scientifiques ont ensuite pu manœuvrer le porte-échantillon pour replacer chaque microcristal d'intérêt cartographié au centre du faisceau de rayons X de précision pour la collecte de données.
Ils ont utilisé l'énergie la plus faible disponible sur la ligne de lumière - réglée pour se rapprocher le plus possible de l'énergie d'absorption des atomes de soufre - et ont collecté des données de diffusion anormale.
"La plupart des lignes de lumière cristallographiques n'ont pas pu atteindre le bord d'absorption de soufre pour des signaux anormaux optimisés, " a déclaré le co-auteur Wayne Hendrickson de l'Université de Columbia. "Heureusement, NSLS-II est une source de lumière synchrotron de premier plan au monde fournissant des rayons X brillants couvrant un large spectre d'énergie de rayons X. Et même si notre niveau d'énergie était légèrement supérieur à l'énergie d'absorption idéale pour le soufre, il a généré les signaux anormaux dont nous avions besoin."
Mais les scientifiques avaient encore du travail à faire pour extraire ces signaux importants et assembler les données de nombreux minuscules cristaux.
« Nous obtenons en fait des milliers de données, " a déclaré Liu. " Nous avons utilisé environ 1400 microcristaux, chacun avec son propre ensemble de données. Nous devons rassembler toutes les données de ces microcristaux."
They also had to weed out data from crystals that were damaged by the intense x-rays or had slight variations in atomic arrangements.
"A single microcrystal does not diffract x-rays sufficiently for structure solution prior to being damaged by the x-rays, " said Sean McSweeney, deputy photon division director and program manager of the Structural Biology Program at NSLS-II. "This is particularly true with crystals of only a few microns, the size of about a bacterial cell. We needed a way to account for that damage and crystal structure variability so it wouldn't skew our results."
They accomplished these goals with a sophisticated multi-step workflow process that sifted through the data, discarded outliers that might have been caused by radiation damage or incompatible crystals, and ultimately extracted the anomalous scattering signals.
"This is a critical step, " said Liu. "We developed a computing procedure to assure that only compatible data were merged in a way to align the individual microcrystals from diffraction patterns. That gave us the required signal-to-noise ratios for structure determination."
Applying the technique
This technique can be used to determine the structure of any protein that has proven hard to crystallize to a large size. These include cell-surface receptors that allow cells of advanced lifeforms such as animals and plants to sense and respond to the environment around them by releasing hormones, transmitting nerve signals, or secreting compounds associated with cell growth and immunity.
"To adapt to the environment through evolution, these proteins are malleable and have lots of non-uniform modifications, " said Liu. "It's hard to get a lot of repeat copies in a crystal because they don't pack well."
In humans, receptors are common targets for drugs, so having knowledge of their varied structures could help guide the development of new, more targeted pharmaceuticals.
But the technique is not restricted to just small crystals.
"The method we developed can handle small protein crystals, but it can also be used for any size protein crystals, any time you need to combine data from more than one sample, " dit Liu.