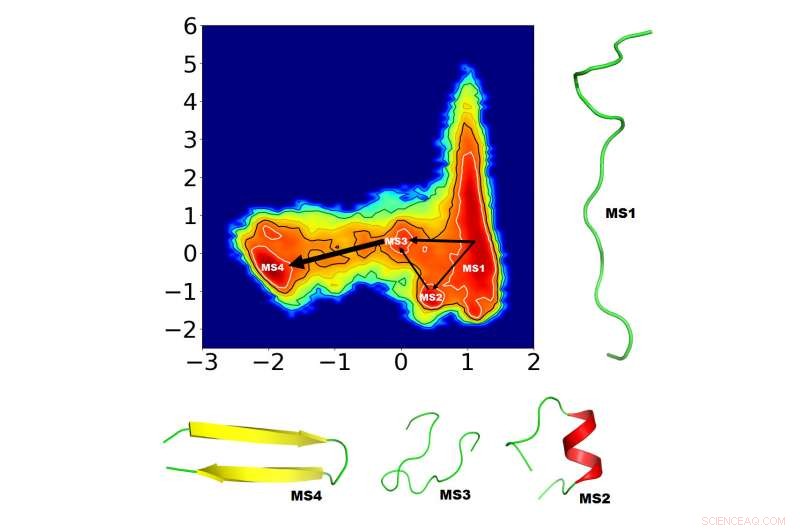
Les scientifiques cherchent à mieux comprendre le repliement des protéines pour guérir les maladies de mauvais repliement, mais ce processus incroyablement complexe nécessite des algorithmes sophistiqués pour identifier les mécanismes de pliage. Les biophysiciens computationnels ont proposé une nouvelle façon d'identifier les facteurs les plus cruciaux pour le repliement des protéines. Ils ont démontré la courte durée de simulation de leur approche sur une petite mais intrigante protéine, "GB1 bêta-épingle à cheveux, " dans le Journal de physique chimique . Les quatre nouveaux états de pliage intermédiaires (MS1-4) identifiés par l'équipe sont présentés ici, ainsi que les voies de connexion possibles. L'épaisseur des flèches d'interconnexion reflète la probabilité que la voie se produise. Crédit :Navjeet Ahalawat et Jagannath Mondal
Les schémas de repliement d'une protéine les aident à effectuer leurs tâches dédiées. En tant que véritables « faiteurs » de la cellule, même une infime altération dans le squelette d'acides aminés d'une protéine peut provoquer un mauvais repliement et entraver la fonctionnalité de la protéine ou provoquer une maladie. Par exemple, si tau, une protéine qui aide à stabiliser la structure des cellules du cerveau, est mal plié, il peut former des tau-enchevêtrements, qui sont fréquemment observées chez les patients atteints de la maladie d'Alzheimer.
Les scientifiques cherchent à mieux comprendre le repliement des protéines pour guérir les maladies de mauvais repliement, mais ce processus incroyablement complexe nécessite des algorithmes sophistiqués pour identifier les mécanismes de pliage. Des biophysiciens computationnels du Tata Institute of Fundamental Research Hyderabad (TIFR-H) ont proposé une nouvelle façon d'identifier les facteurs les plus cruciaux pour le repliement des protéines. Ils ont démontré la courte durée de simulation de leur approche sur une petite mais intrigante protéine, "GB1 bêta-épingle à cheveux, " dans le Journal de physique chimique , des éditions AIP.
"En combinant une méthode connue sous le nom d'analyse des composants indépendants basée sur la structure temporelle (TICA) avec de courtes simulations de dynamique moléculaire, nous avons trouvé quatre états de pliage intermédiaires physiquement significatifs, non observé auparavant, et a montré des états hélicoïdaux qui ne peuvent généralement pas être détectés par d'autres méthodes, " a déclaré Navjeet Ahalawat, un auteur sur le papier.
Chaque atome d'une protéine peut se replier en trois dimensions, mais avec des millions d'atomes présents même dans des protéines simples, la tâche de comprendre la combinaison de pliage collective devient alambiquée. Les scientifiques ont examiné les différents facteurs influençant le repliement des protéines, telles que les liaisons hydrogène, et combinés dans des descriptions générales appelées variables collectives (CV). Cependant, avec beaucoup de facteurs potentiels, les scientifiques ne disposent pas d'un bon moyen de trouver des CV qui décrivent de manière appropriée un processus réalisable.
"Il existe de nombreuses façons pour les protéines de passer de l'état déplié à l'état replié, donc le plus difficile est de décider par où commencer, " Ahalawat a dit. Jagannath Mondal, un autre auteur sur le papier, a ajouté qu'il était facile de "se perdre dans les données".
L'équipe a décidé d'étudier l'épingle à cheveux en saillie externe de la protéine GB1 en raison du grand nombre de travaux existants et des nombreuses possibilités de repliement potentielles déjà estimées dans les CV antérieurs. Ahalawat et Mondal ont pris un certain nombre de CV GB1 existants comme CV constitutifs et les ont combinés linéairement à l'aide de TICA pour identifier une paire de CV « optimisés ». Puis, ils ont saisi les CV optimisés dans le modèle d'état de Markov et identifié quatre états de repliement intermédiaires ainsi que les voies de connexion possibles.
"Nous avons demandé, quelles sont les caractéristiques estimées précédemment pour cette protéine particulière qui pourraient vraiment jouer un rôle clé dans le système ? Et pouvons-nous trouver la bonne combinaison de conditions ?", A déclaré Ahalawat. « Dans notre travail, nous pouvons maintenant déterminer quantitativement si cette caractéristique est pertinente pour le processus. »
« À l'aide de courtes simulations, nous avons trouvé le poids que vous avez vraiment besoin d'utiliser dans une combinaison, et cela donne le bon modèle de pliage pour une protéine, " Mondal a ajouté. " C'est un moyen vraiment bon marché de comprendre le repliement des protéines. "
Dans leur méthode, les données d'études antérieures sont nécessaires pour identifier les CV optimaux. L'équipe envisage que leur technique puisse être utilisée pour découvrir le mécanisme interne du repliement sain des protéines afin de corriger la maladie causant des protéines mal repliées. Ils souhaitent également développer davantage leur méthode d'optimisation du CV et l'appliquer à la reconnaissance biomoléculaire et à la découverte de médicaments. « À l'avenir, nous prévoyons d'intégrer des méthodes non linéaires, en utilisant des techniques d'apprentissage en profondeur basées sur les réseaux de neurones pour améliorer notre modèle, " a déclaré Ahalawat.