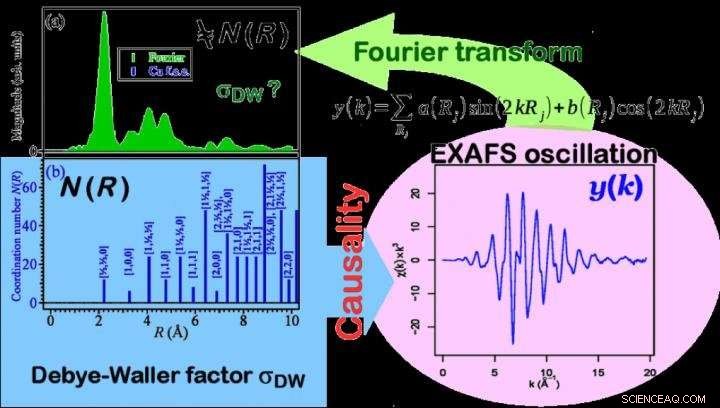
Les lignes bleues de la figure (b) décrivent la fonction de structure radiale correcte, N(R), tel que déterminé par la structure du matériau. Le N(R) a des composantes radiales (le nombre d'atomes de coordination) à faible distance interatomique R. Au fur et à mesure que la distance augmente, le N(R) augmente le nombre d'atomes (intensité de N(R)) se coordonnant à la même distance en raison de la symétrie structurelle des substances. Contrairement à un tel N(R) discret, nous avons obtenu des informations sur la microstructure à partir d'un spectre vert sur la figure (a), qui est calculé par transformation de Fourier du spectre d'oscillation EXAFS (côté droit de la figure). Ce spectre est une fonction continue de la distance interatomique R, et montre que l'intensité diminue à mesure que R augmente. Cela signifie que lorsque la distance atomique augmente, cette méthode ne peut pas estimer correctement la fonction de structure radiale des atomes de coordination. Crédit :Dr Ichiro Akai
L'analyse de la microstructure des matériaux est une technologie clé pour la recherche de nouveaux matériaux. En utilisant une technique d'extraction d'informations appelée modélisation creuse, une collaboration de chercheurs dirigée par le professeur Ichiro Akai de l'Université de Kumamoto, Japon, a développé la première méthode au monde d'analyse de la structure atomique et des fluctuations structurelles des matériaux en utilisant uniquement des données mesurées. Cette méthode ne nécessite aucune hypothèse préalable sur la structure à l'échelle atomique, qui sont nécessaires dans les méthodes conventionnelles d'analyse de la microstructure. Entre autres applications, cette nouvelle approche devrait améliorer la fonctionnalité des batteries et prolonger leur durée de vie.
Pour réaliser de nouvelles fonctions et améliorations des performances des substances fonctionnelles telles que celles trouvées dans les batteries et les appareils électroniques, leur structure et leurs changements structurels doivent être évalués à l'échelle atomique. En effet, la structure des atomes à l'échelle nanométrique domine leurs propriétés. Les mesures EXAFS (Extended X-ray Absorption Fine Structure) sont largement utilisées pour analyser de telles microstructures à l'échelle atomique.
En effectuant une transformation de Fourier sur le spectre mesuré d'une oscillation EXAFS, des informations sur la microstructure peuvent être obtenues pour déterminer comment les atomes adjacents sont distribués radialement. Cependant, la distribution radiale obtenue par cette méthode classique est assez différente de la structure radiale réelle. Cet écart est dû à une expansion incorrecte avec des fonctions de base d'ondes vibrantes d'amplitude constante par transformation de Fourier, malgré le fait que l'amplitude de l'oscillation EXAFS change sensiblement dans la plage observée.
Les changements d'amplitude représentent des fluctuations structurelles, qui sont les variations des distances atomiques et la mobilité des atomes voisins. Ces propriétés physiques sont indiquées par une quantité physique appelée facteur Debye-Waller. Ce facteur ne peut pas être obtenu par transformation de Fourier de l'oscillation EXAFS car l'estimation du facteur Debye-Waller nécessite de faire des hypothèses sur la microstructure d'un matériau. En d'autres termes, puisqu'une analyse du spectre d'oscillation EXAFS conventionnel est basée sur une structure hypothétique, il est difficile d'estimer le facteur Debye-Waller à moins que la microstructure du matériau ne soit préalablement connue.
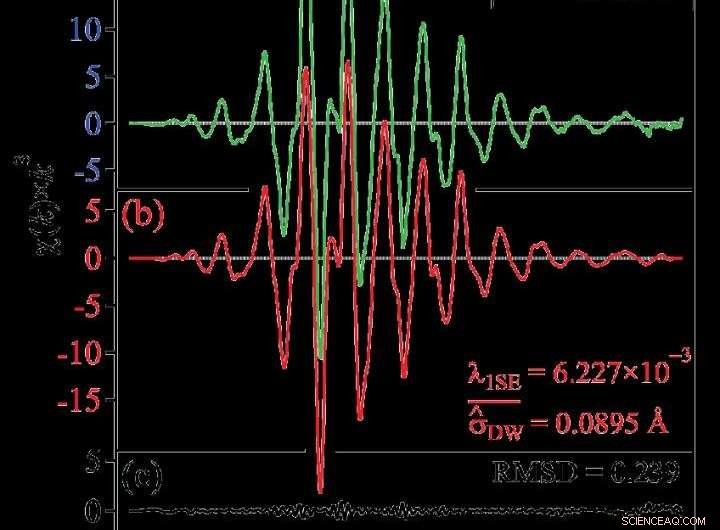
(a) Données mesurées, (b) Résultat après modélisation creuse, (c) Résidu du résultat par modélisation éparse des données mesurées. Crédit :Dr Ichiro Akai
Pour résoudre ce problème, les chercheurs se sont concentrés sur le fait que les atomes sont, en général, distribué régulièrement, qui reflète leur structure chimique et leurs états de liaison. Par ailleurs, les distances entre atomes (coordonnées atomiques) sont distinctes, et peut être considéré comme « clairsemé ». Les chercheurs ont ensuite développé une nouvelle méthode analytique utilisant un type de technologie d'extraction d'informations appelée modélisation éparse pour analyser les données EXAFS. La modélisation creuse a été développée dans le domaine des sciences de l'information, et est utilisé pour découvrir les propriétés dominantes dans les données mesurées. Dans les années récentes, il a été utilisé dans un large éventail de domaines de recherche, comme l'astronomie, sciences médicales et ingénierie.
En utilisant uniquement des données mesurées, sans aucune connaissance préalable d'un matériau, la nouvelle méthode peut
"Comme nous pouvons estimer le facteur Debye-Waller sans aucune information préalable sur un matériau, nous nous attendons à ce que cette méthode produise des résultats importants dans plusieurs domaines de la recherche sur les matériaux, en particulier pour les nouvelles substances, tels que les matériaux thermoélectriques où la fluctuation thermique des atomes adjacents est importante, et les matériaux conducteurs superioniques qui nécessitent une mobilité entre des atomes adjacents. Ces deux éléments attirent actuellement l'attention en tant que matériaux d'électrolyte solide pour les batteries secondaires, " a déclaré le chercheur principal, Professeur Ichiro Akai de l'Université de Kumamoto.
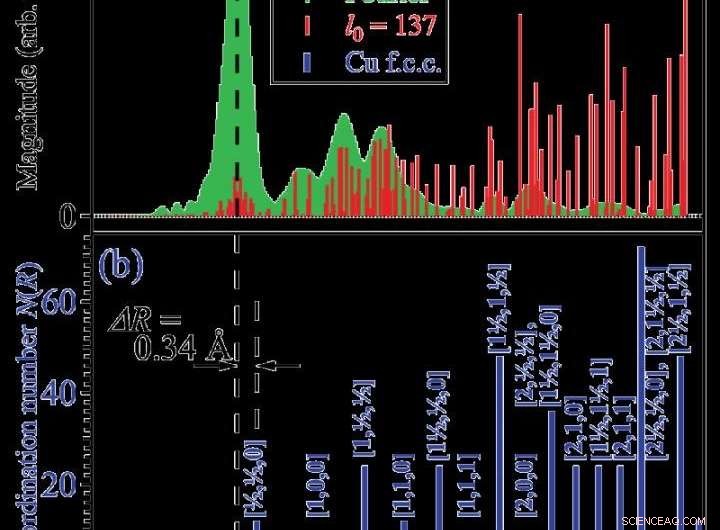
(a) Vert :spectre de transformation de Fourier classique. Rouge :fonction de structure quasi-radiale extraite par modélisation creuse. (b) Fonction de structure radiale correcte du cuivre. Crédit :Dr Ichiro Akai
Dans cette étude, les chercheurs ont appliqué leur nouvelle méthode aux données EXAFS à partir d'un échantillon standard de cuivre et ont démontré que la technique de modélisation éparse fonctionnait correctement et efficacement pour analyser le spectre d'oscillation EXAFS. L'application de cette méthode à divers matériaux difficiles à analyser en détail par des méthodes conventionnelles devrait produire des développements futurs.
Ce travail a été publié dans le Journal de la Société de physique du Japon le 22 juin 2018.