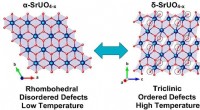
Modèle d'un AgONist clivable par réduction (RECON) pour le contrôle réversible des voies de signalisation cellulaire dépendantes du GPCR. Crédit :J. Broichhagen, D. Hodson
Les chercheurs du LMU ont développé une méthode qui permet d'activer et de désactiver à volonté les récepteurs de la surface cellulaire. La technique promet de fournir de nouvelles informations sur les fonctions des récepteurs et leurs effets sur les voies de signalisation intracellulaires.
Les récepteurs transmembranaires couplés aux protéines G (RCPG) sont intimement impliqués dans la régulation d'un large éventail de processus biologiques. Ils fonctionnent en liant des molécules messagères extracellulaires - telles que des hormones - et en activant des relais de signalisation intracellulaires qui modifient les fonctions cellulaires de manière spécifique. Sans surprise, les défauts fonctionnels de ces récepteurs liés à la membrane conduisent souvent à des troubles physiologiques chroniques. Cela explique pourquoi une grande partie de la recherche pharmacologique actuelle se concentre sur la recherche de médicaments qui ciblent des GPCR spécifiques. Une collaboration de recherche impliquant des groupes dirigés par les professeurs Anja Hoffmann-Röder (professeur de chimie organique) au LMU, Dirk Trauner (anciennement LMU, maintenant à l'Université de New York) et David Hodson (Université de Birmingham) a maintenant conçu une suite de molécules avec lesquelles les GPCR (et potentiellement d'autres types de récepteurs) peuvent être activés et désactivés pharmacologiquement à volonté. Ces agents promettent de découvrir de nouveaux aspects des mécanismes moléculaires qui sous-tendent les actions des récepteurs, et, ce faisant, devrait faciliter le développement de nouvelles stratégies thérapeutiques. Les nouvelles découvertes apparaissent dans le journal en ligne ACS Science centrale .
La nouvelle étude est basée sur des travaux antérieurs dans lesquels l'équipe avait développé un système pharmacologique dépendant de la lumière à l'aide de « SNAP-tags ». Un SNAP-tag est une protéine de liaison qui peut être introduite dans une protéine réceptrice cible par des moyens génétiques. Son but est d'agir comme un site de liaison pour la fixation d'un ligand synthétique, qui peut alors moduler la signalisation des récepteurs, c'est-à-dire activer ou bloquer l'activation. Le nouvel article décrit l'application de cette technique à un récepteur appelé GLP-1R, qui régule la sécrétion d'insuline. Il présente donc une cible intéressante pour le développement de médicaments permettant de traiter le diabète. "Nous avons utilisé comme ligand une hormone naturelle qui était équipée d'une extension synthétique qui agit comme un adaptateur qui est lié de manière covalente par le SNAP-tag. La liaison de l'hormone active le récepteur, tandis que la fixation directe de l'adaptateur au SNAP-tag garantit que le récepteur est maintenu dans l'état ON, " explique Tom Podewin (Max Planck Institute for Medical Research), co-premier auteur de la nouvelle étude et jusqu'en 2017, doctorant dans le groupe Hoffmann-Röder. La fixation de l'extrémité de l'adaptateur au SNAP-tag attache efficacement le ligand au récepteur. Cependant, l'adaptateur est relié de manière flexible au site de liaison aux hormones du récepteur via une liaison disulfure, qui peut être facilement rompu par l'ajout d'un agent réducteur. Cette astuce permet à l'hormone d'être libérée de son site de liaison, inversant ainsi l'interaction et éteignant à nouveau le récepteur.
Afin de démontrer la polyvalence de cette approche de « pharmacologie captive », l'équipe a utilisé un ligand synthétique différent pour activer un récepteur qui contrôle la sécrétion de l'hormone de croissance. "Nos ligands sont en fait les plus grandes molécules captives connues qui se sont avérées agir en tant qu'activateurs ou agonistes des récepteurs liés à la membrane. Cela prouve que la pharmacologie captive ne se limite pas à l'utilisation de petites molécules, mais peut être étendu aux peptides et peut-être même aux protéines, ", souligne Hoffmann-Röder.
Étant donné que la liaison au SNAP-tag est covalente, le ligand activateur ne peut pas être facilement libéré du récepteur en l'absence d'un agent réducteur. Normalement, Les GPCR activés sont rapidement retirés de la membrane cellulaire et transportés vers les vésicules intracellulaires. Une fois là, leurs ligands se dissocient et ils sont ensuite recyclés à la surface cellulaire. Cependant, les chercheurs ont découvert - à leur grande surprise - que la liaison du ligand synthétique inhibe ce processus de recyclage, piégeant le récepteur dans la vésicule. "La capacité d'attacher durablement n'importe quel ligand - qu'il s'agisse d'un agent pharmacologique ou d'un marqueur à utiliser en bio-imagerie - à un récepteur convenablement modifié offre de nouvelles opportunités pour la manipulation et la caractérisation de voies de signalisation complexes dans les cellules, " ajoute le premier auteur conjoint Johannes Broichhagen. Lui et ses collègues pensent que la nouvelle méthode permettra de mieux comprendre les récepteurs et leurs fonctions, qui aura sans aucun doute des répercussions sur le développement de médicaments à l'avenir.