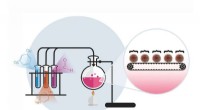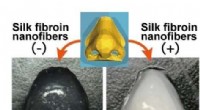
Principe de la cristallographie synchrotron série mix-and-diffuse :les cristaux de protéines sont mélangés avec une solution d'un candidat médicament et radiographiés sur une bande passant dans le faisceau de rayons X. Crédit :Beyerlein et al., IUCrJ
Les scientifiques de DESY ont développé une nouvelle méthode qui permet un criblage automatisé et rapide de candidats-médicaments prometteurs. Cette nouvelle technique, appelée cristallographie synchrotron série mix-and-diffuse, peut imager l'interaction de cibles médicamenteuses potentielles avec des médicaments candidats ou d'autres molécules. Le concept a le potentiel de porter la structure et la conception de médicaments à base de fragments à un nouveau niveau, comme l'écrivent les chercheurs dans le Journal de l'Union Internationale de Cristallographie ( IUCrJ ).
De nombreuses protéines dans le corps sont des cibles potentielles de médicaments. Des molécules pharmaceutiques de forme correcte peuvent se lier à ces protéines et activer ou désactiver leur fonction. Par exemple, pour lutter contre certaines formes de leucémie, le médicament anticancéreux Imatinib inhibe une variante hyperactive de l'enzyme tyrosine-kinase, une protéine responsable de l'activation de nombreuses autres protéines. L'imatinib bloque le site actif de cette tyrosine-kinase. Pour y parvenir, la molécule de médicament doit s'insérer précisément dans le site actif comme une clé dans une serrure. Sur la base de la connaissance de la structure spatiale de l'enzyme cible, L'imatinib a été conçu sur mesure à cet effet.
"Cette stratégie est appelée conception de médicaments basée sur la structure et est aujourd'hui utilisée comme méthode standard dans le développement de médicaments pharmaceutiques, " explique le premier auteur Kenneth Beyerlein du Center for Free-Electron Laser Science (CFEL), une coopération de DESY, l'Université de Hambourg et la Société allemande Max Planck. "Toutefois, en réalité, cibler des protéines est beaucoup plus complexe que d'insérer une clé dans une serrure. Par conséquent, de nombreuses molécules pharmaceutiques potentielles ou fragments de telles molécules doivent être testés, ce qui est généralement une procédure longue et compliquée. les biologistes et les pharmacologues s'intéressent au fonctionnement précis des agents naturels qui se lient aux protéines, pour mieux comprendre les rouages de la vie.
Le système développé par l'équipe autour de Beyerlein et son collègue de DESY Dominik Oberthür, aussi du CFEL, offre une nouvelle façon de poursuivre cet objectif :il mélange des protéines microcristallines avec des molécules spécifiques appelées ligands qui peuvent être des candidats-médicaments ou des agents naturels juste avant de sonder les cristaux avec des rayons X pour révéler la structure spatiale détaillée du complexe protéine-ligand résultant ou le absence d'un tel complexe si un ligand potentiel ne se lie pas à la protéine.
Pour analyser la structure spatiale d'une protéine, les scientifiques utilisent souvent la cristallographie aux rayons X. Pour cette technique, un cristal doit d'abord être développé à partir de la protéine. Les chercheurs prennent ensuite des clichés aux rayons X de tous les côtés du cristal qui doit être refroidi à des températures ultra-basses pour réduire les dommages causés par le rayonnement intense. Les rayons X produisent un diagramme de diffraction caractéristique à partir duquel la structure interne du cristal et donc la structure spatiale de la protéine peuvent être calculées. Pour étudier une protéine avec un ligand, un nouveau cristal doit être cultivé à partir d'une solution de protéine et de ligand ou le cristal doit être imbibé de ligand. Même avec l'utilisation de la robotique pour automatiser toutes les étapes de ce processus, la nécessité de monter des cristaux individuels pour chaque nouvel ensemble de données est devenue l'étape limitante dans le criblage de grandes bibliothèques de composés.
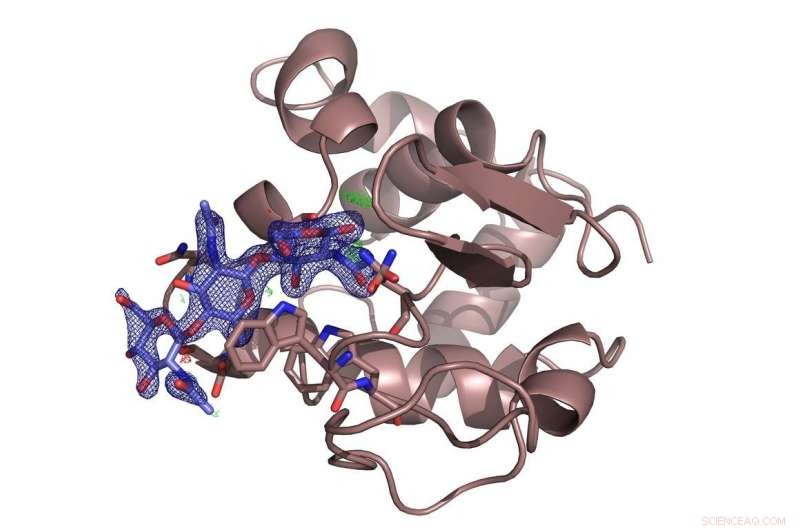
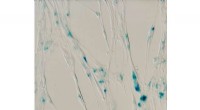
L'enzyme lysozyme (brun) avec le sucre inhibiteur chitotriose (bleu) lié à celui-ci. L'enquête a réglé une controverse sur le site de liaison préféré de la molécule de sucre. Crédit :DESY, Dominik Oberthür
La nouvelle technique suit une approche différente. "Nous utilisons des microcristaux qui ont deux avantages :ils sont généralement beaucoup plus faciles à produire que les gros cristaux, et ils sont suffisamment petits pour qu'un médicament potentiel dans une solution puisse diffuser à travers le cristal et se lier à toutes les molécules de protéines en quelques millisecondes, " explique Oberthür. Le système développé par l'équipe d'Oberthür et de Beyerlein distribue un flux de microcristaux dans un liquide porteur sur une fine bande. Comme une bande transporteuse, la bande transporte les cristaux à travers le faisceau de rayons X, qui est découpé en brefs éclairs par un store rotatif. Au lieu de faire tourner un gros cristal dans le faisceau de rayons X, de nombreux microcristaux en orientation aléatoire sont ainsi soumis à des rayons X en série et les diagrammes de diffraction de chaque tir sont ensuite combinés pour former un ensemble de données complet, suivant le concept de cristallographie en série qui a d'abord été développé dans les lasers à rayons X à électrons libres (XFEL).
Grâce à une deuxième vanne dans le système, une solution d'un candidat médicament ou d'un ligand naturel est ajoutée. Le point où les deux liquides se mélangent peut être ajusté pour créer un délai défini avant d'étudier la structure. Cette configuration ne nécessite pas de cryo-refroidissement des cristaux, donc l'interaction protéine-médicament peut être observée à des températures physiologiques, ou toute autre température souhaitée. Par ici, même la dynamique de liaison peut être étudiée. "Nous pouvons diffuser des produits chimiques dans les cristaux de protéines à la volée et regarder la liaison se produire, " explique Oberthür. " Vous n'avez pas à trouver de nouvelles conditions de croissance pour chaque inhibiteur et vous n'avez pas besoin d'échanger les cristaux manuellement, l'ensemble du processus peut être automatisé."
L'équipe a testé le nouveau système sur la source de rayons X à haute brillance PETRA III de DESY avec la protéine bien connue lysozyme et une molécule de sucre, chitotriose, qui inhibe l'enzyme. Les microcristaux de lysozyme utilisés ici n'avaient qu'environ six à huit micromètres de diamètre. L'installation à la station de mesure P11 a révélé en détail la structure spatiale de l'inhibiteur mélangé lié au lysozyme. Et même si la structure du lysozyme a été la première structure enzymatique révélée par cristallographie aux rayons X il y a 50 ans, la nouvelle méthode pourrait encore révéler de nouveaux détails sur le mode de liaison du chitotriose au lysozyme, régler une controverse sur le site de liaison préféré de la molécule de sucre.
Alors que la preuve de principe demandait encore du temps, Les progrès de routine et ultérieurs des détecteurs et de la technologie des rayons X accéléreront considérablement la procédure. Aussi, en utilisant tout le spectre du faisceau de rayons X de la source lumineuse synchrotron au lieu d'une seule "couleur" de celle-ci, peut pousser le temps d'exposition pour les images de diffraction individuelles jusqu'à 100 picosecondes, ou 0,1 milliardième de seconde. Seulement 50 de ces images sont suffisantes pour déterminer la structure, comme cela a été montré récemment.
« Nous développons des moyens de résoudre la structure des protéines liées pour la découverte de médicaments à haut débit, " explique Beyerlein. Comme les sources lumineuses synchrotron sont plus accessibles que les lasers à rayons X, les chercheurs imaginent d'utiliser cette méthode pour le criblage de routine grâce à des bibliothèques d'inhibiteurs potentiels et de fragments de médicaments. « Le faire automatiquement et beaucoup plus rapidement qu'avec les approches conventionnelles serait un grand pas en avant dans la conception de médicaments basés sur la structure, " dit Beyerlein.