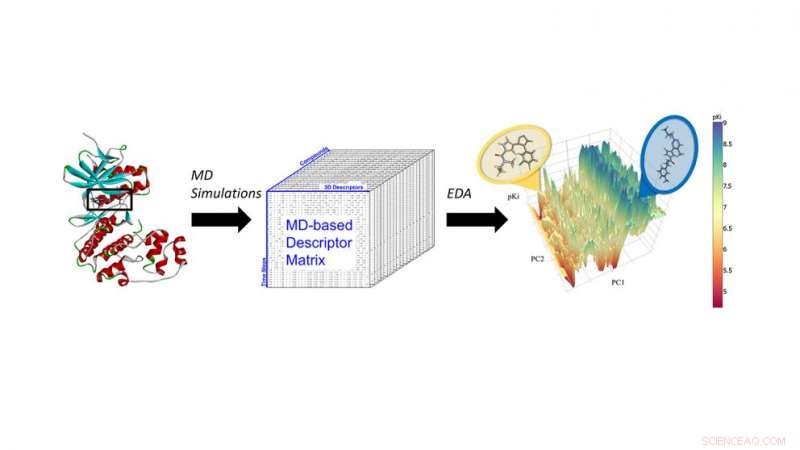
Simulations de dynamique moléculaire (MD) d'inhibiteurs d'ERK2 pour extraire des descripteurs MD pour l'analyse chimico-informatique de nouvelle génération et l'apprentissage automatique. Crédit :Université d'État de Caroline du Nord
Des chercheurs de la North Carolina State University ont démontré que les simulations de dynamique moléculaire et les techniques d'apprentissage automatique pouvaient être intégrées pour créer des modèles de prédiction informatique plus précis. Ces modèles « hyper-prédictifs » pourraient être utilisés pour prédire rapidement quels nouveaux composés chimiques pourraient être des candidats médicaments prometteurs.
Le développement de médicaments est un processus long et coûteux. Pour réduire le nombre de composés chimiques qui pourraient être des candidats médicaments potentiels, les scientifiques utilisent des modèles informatiques qui peuvent prédire comment un composé chimique particulier pourrait interagir avec une cible biologique d'intérêt - par exemple, une protéine clé qui pourrait être impliquée dans un processus pathologique. Traditionnellement, cela se fait via la modélisation quantitative de la relation structure-activité (QSAR) et l'amarrage moléculaire, qui reposent sur des informations 2 et 3D sur ces produits chimiques.
Denis Fourches, professeur assistant de chimie computationnelle, voulait améliorer la précision de ces modèles QSAR. « Lorsque vous examinez un ensemble de 30 millions de composés, vous n'avez pas nécessairement besoin d'une très grande fiabilité avec votre modèle - vous n'avez qu'une idée approximative des 5 ou 10 % les plus importants de cette bibliothèque virtuelle. Mais si vous essayez de réduire un champ de 200 analogues à 10, ce qui est plus souvent le cas dans le développement de médicaments, votre technique de modélisation doit être extrêmement précise. Les techniques actuelles ne sont certainement pas assez fiables."
Fourches et Jérémy Ash, un étudiant diplômé en bioinformatique, a décidé d'incorporer les résultats des calculs de dynamique moléculaire - des simulations de tous les atomes de la façon dont un composé particulier se déplace dans la poche de liaison d'une protéine - dans des modèles de prédiction basés sur l'apprentissage automatique.
"La plupart des modèles n'utilisent que les structures bidimensionnelles des molécules, " dit Fourches. " Mais en réalité, les produits chimiques sont des objets tridimensionnels complexes qui se déplacent, vibrent et ont des interactions intermoléculaires dynamiques avec la protéine une fois ancrée dans son site de liaison. Vous ne pouvez pas voir cela si vous regardez simplement la structure 2-D ou 3-D d'une molécule donnée."
Dans une étude de validation de principe, Fourches et Ash ont étudié la kinase ERK2 - une enzyme associée à plusieurs types de cancer - et un groupe de 87 inhibiteurs connus d'ERK2, allant de très actif à inactif. Ils ont effectué des simulations indépendantes de dynamique moléculaire (MD) pour chacun de ces 87 composés et ont calculé des informations critiques sur la flexibilité de chaque composé une fois dans la poche ERK2. Ensuite, ils ont analysé les descripteurs MD à l'aide de techniques de chimioinformatique et d'apprentissage automatique. Les descripteurs MD ont permis de distinguer avec précision les inhibiteurs actifs d'ERK2 des actifs faiblement et inactifs, ce qui n'était pas le cas lorsque les modèles n'utilisaient que des informations structurelles 2D et 3D.
"Nous avions déjà des données sur ces 87 molécules et leur activité à ERK2, " dit Fourches. " Nous avons donc testé pour voir si notre modèle était capable de trouver de manière fiable les composés les plus actifs. En effet, il distinguait avec précision les inhibiteurs ERK2 forts et faibles, et parce que les descripteurs MD ont codé les interactions que ces composés créent dans la poche d'ERK2, cela nous a également permis de mieux comprendre pourquoi les inhibiteurs puissants fonctionnaient bien.
« Avant que les progrès de l'informatique ne permettaient de simuler ce type de données, il nous aurait fallu six mois pour simuler une seule molécule dans la poche d'ERK2. Grâce à l'accélération GPU, maintenant cela ne prend que trois heures. C'est un changeur de jeu. J'espère que l'incorporation de données extraites de la dynamique moléculaire dans les modèles QSAR permettra une nouvelle génération de modèles hyper-prédictifs qui aideront à apporter de nouveaux, médicaments efficaces sur le marché encore plus rapidement. C'est l'intelligence artificielle qui travaille pour nous pour découvrir les médicaments de demain. »