Les technologies de séquençage de l'ADN de nouvelle génération ont inondé les bases de données et les disques durs du monde entier avec de grands ensembles de données, mais les chercheurs tirent-ils le meilleur parti de ce déluge de données ? Dans une nouvelle étude publiée dans le numéro d'octobre de Applications en sciences végétales , Le Dr Brent Berger et ses collègues proposent une façon de tamiser l'or restant à partir de grands ensembles de données de séquence. Les auteurs montrent qu'une nouvelle technique d'exploration de données peut être utilisée pour glaner des informations précieuses à partir d'ensembles de données existants, et prouver le concept en récupérant la séquence de gènes influençant les structures florales particulières observées dans la famille de plantes Goodeniaceae.
Le séquençage de l'ADN est devenu si bon marché que même si un chercheur ne s'intéresse vraiment qu'à la séquence de quelques gènes, il est souvent plus pratique de simplement séquencer le génome entier. Les techniques bioinformatiques peuvent sélectionner la séquence génétique souhaitée plus tard, avec moins de tracas que de cibler des gènes spécifiques à séquencer. Cette pratique, connu sous le nom d'« écrémage du génome », " est devenu un moyen de plus en plus populaire de répondre aux questions sur les relations entre les espèces végétales.
La prémisse de l'écrémage du génome est d'utiliser le séquençage au fusil de chasse à faible couverture pour récupérer la séquence d'ADN à partir de fractions à copie élevée du génome. Dans le séquençage du fusil de chasse, le génome est divisé en petits morceaux pour le séquençage, puis recousus par calcul en utilisant les chevauchements entre les morceaux, un processus appelé assemblage. La quantité de « couverture » correspond au nombre de ces petits morceaux séquencés ; plus la couverture est élevée, plus il est facile de recoudre le génome, résultant en une séquence génomique plus complète.
Mais une couverture plus élevée coûte plus cher, et certaines questions peuvent être répondues avec un moins cher, cycle de séquençage à faible couverture. « fractions à forte copie » de l'ADN génomique total, tels que les génomes chloroplastiques ou l'ADN ribosomique nucléaire, sont en plus grande abondance dans le pool de séquences, et peut donc être entièrement séquencé même en pas cher, pistes à faible couverture. Les séquences de ces fractions génomiques à copie élevée sont généralement utilisées pour résoudre les relations évolutives entre différentes espèces et groupes. Mais dans le processus d'écrémage du génome, les chercheurs produisent puis rejettent d'énormes quantités de données de séquence potentiellement précieuses. "De nombreux ensembles de données d'écrémage du génome sont utilisés pour assembler le génome du chloroplaste, qui dans notre cas, n'a utilisé que 3% des données séquencées, " a fait remarquer le Dr Dianella Howarth, un co-auteur de l'étude.
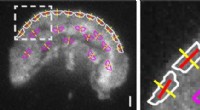
Dans cette étude, les auteurs ont examiné à nouveau un ensemble de données d'écrémage du génome précédemment utilisé pour résoudre les relations évolutives chez les Goodeniaceae, une famille de plantes communément appelées « fleurs en éventail » ou « demi-fleurs » en raison de leur forme de fleur intrigante, on dirait que quelqu'un a coupé la fleur en deux. Les auteurs voulaient voir si cet ensemble de données d'écrémage du génome pouvait être sondé pour plus d'informations sur la génétique derrière cette structure florale unique. Ils ont utilisé plusieurs progiciels pour assembler des fragments de séquence inutilisés auparavant à partir de la fraction à faible nombre de copies de l'ensemble de données d'écrémage du génome d'origine. Ils ont ensuite recherché dans l'assemblage résultant la séquence d'un ensemble de gènes appelés CYCLOIDEE gènes, qui sont impliqués dans la structure florale et la symétrie.
Les auteurs ont pu récupérer suffisamment de portions des gènes, de plusieurs espèces, pour créer des alignements complets des quatre CYCLOIDEE gènes dans le noyau Goodeniaceae. Ces données pourraient s'avérer utiles pour de futures études sur l'évolution de la structure florale bizarre observée dans ce groupe. "Comparer des séquences de CYCLOIDEE -les gènes similaires à travers ce clade pourraient fournir des indices sur les changements de séquence précis qui entraînent des changements dans la morphologie florale, " a expliqué le Dr Howarth.
Plus généralement, Le Dr Howarth a continué, "Des morceaux de tout gène d'intérêt pourraient potentiellement être extraits d'ensembles de données d'écrémage du génome qui ont déjà été complétés." Un morceau de gène peut sembler peu, mais il y a un nombre surprenant d'utilisations pour ces fragments. "Ces données pourraient fournir suffisamment d'informations pour déterminer les régions nucléaires utiles pour les analyses phylogénétiques ou identifier d'éventuels événements de duplication de gènes. De plus, des sondes pour le séquençage d'enrichissement de cibles pourraient être générées rapidement à travers un clade pour examiner les gènes candidats et leurs régions régulatrices dans des études evo-devo. »
Des approches d'exploration de données comme celles-ci permettent une utilisation beaucoup plus complète des ensembles de données d'écrémage du génome. Cela permet de répondre à des questions importantes avec les données existantes, et ouvre la porte aux scientifiques sans accès aux ressources pour produire des ensembles de données à grande échelle, par exemple, scientifiques dans des collèges plus petits ou dans des pays sans grands organismes subventionnaires. Alors que les données sur les séquences d'ADN continuent d'affluer, des études comme celle-ci indiquent des moyens de s'assurer que nous ne laissons pas passer des informations précieuses.