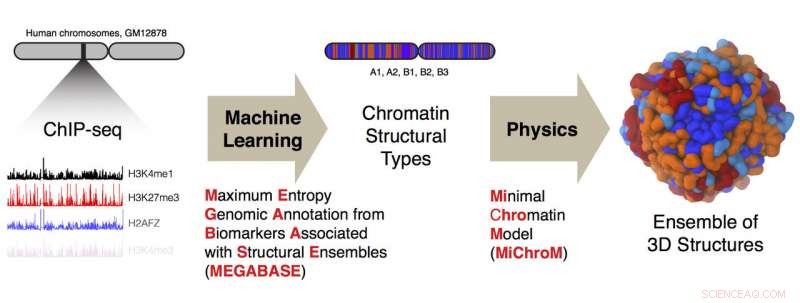
Des chercheurs de l'Université Rice et du Baylor College of Medicine ont développé un pipeline informatique pour convertir les données de séquençage de ChIP unidimensionnel sur l'ADN en structures tridimensionnelles de chromosomes humains. Crédit :Ryan Cheng/Michele Di Pierro
L'ADN d'une cellule humaine mesure 1,83 mètre de long et s'enroule autour de millions de protéines histones semblables à des billes pour s'adapter à l'intérieur du noyau de la cellule. Des chercheurs de l'Université Rice et du Baylor College of Medicine ont montré que l'examen de l'état chimique de ces protéines permet de prédire comment un chromosome d'ADN entier va se replier.
Des chercheurs basés au Centre de physique biologique théorique (CTBP) de Rice ont construit des modèles informatiques pour analyser les marques épigénétiques, qui comprennent des protéines liées à l'ADN ainsi que des modifications chimiques des protéines histones. Ils ont récolté les informations codées dans ces marquages pour prédire comment les chromosomes se replient en trois dimensions.
Leurs découvertes rapprochent le domaine de la génétique de la capacité de prédire la structure repliée de génomes entiers, qui pourrait un jour aider à identifier les maladies génétiques liées à un mauvais repliement.
L'ouvrage paraît cette semaine dans le Actes de l'Académie nationale des sciences .
Emballé dans le noyau, L'ADN se replie sous une forme fonctionnelle qui diffère selon les différents types de cellules. Parce que chaque cellule d'un organisme contient le même ADN, les marques épigénétiques l'aident à trouver la bonne forme pour le type de cellule qu'elle habite.
"Quelque chose au-dessus du code génétique dit à la cellule ce qu'elle est censée être et détermine quelles parties du chromosome vont être lues à un moment donné, " a déclaré le biophysicien Peter Wolynes, un co-auteur de l'article. "Ce sont les soi-disant marques épigénétiques."
Collectivement, les marques épigénétiques aident à emballer le génome dans les compartiments lâches mais très organisés qu'il adopte pendant l'interphase, le "moyen âge" de travail dans la vie d'une cellule. Ces compartiments rapprochent étroitement les gènes liés à la transcription et leur permettent de communiquer et de fonctionner.
Les marques épigénétiques peuvent être révélées par une technique établie appelée séquençage ChIP, qui cartographie les sites de liaison aux protéines le long de l'ADN.
"Nous ne comprenons pas exactement comment le génome est marqué, mais nous pouvons le mesurer grâce au séquençage ChIP, qui est devenue une expérience assez simple, " a déclaré Wolynes. " De la même manière que nous pouvons voir le code génétique (l'ADN), nous pouvons également mesurer ces marques directement dans de nombreuses cellules différentes. Ils sont devenus la prochaine couche de séquence sur le génome."
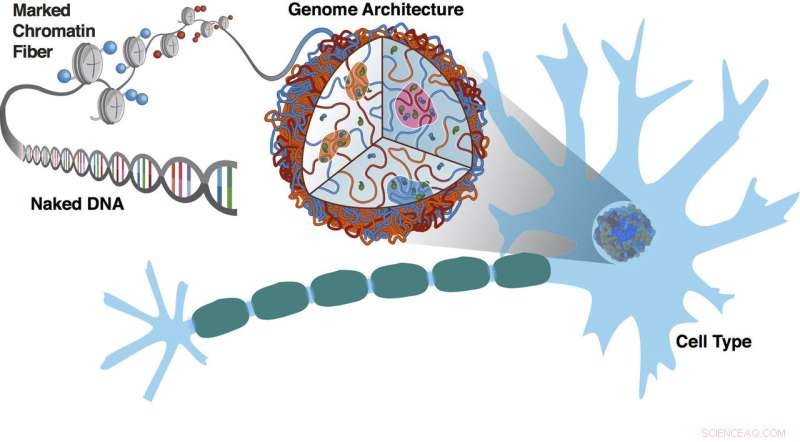
Des expériences à l'Université Rice et au Baylor College of Medicine montrent comment des segments de chromatine avec les mêmes motifs de marquage épigénétique se localisent ensemble dans un processus lié à la séparation de phases. L'ADN nu est décoré par des marques épigénétiques qui codent l'arrangement tridimensionnel des chromosomes. L'architecture du génome et les motifs de marquage sont des caractéristiques du type cellulaire, dans ce cas une cellule nerveuse avec sa gaine de myéline caractéristique. Crédit :Sigrid Knemeyer/Center for Theoretical Biological Physics at Rice University
"C'est un autre niveau d'information, " a déclaré le co-auteur et biophysicien José Onuchic. "Chacun de l'ADN de vos cellules est le même. Cependant, différents types de cellules ont une épigénétique différente, donc leurs modèles d'expression sont différents."
Les co-auteurs principaux et les boursiers postdoctoraux Rice Michele Di Pierro et Ryan Cheng ont utilisé des données de séquençage ChIP pour une cellule lymphoblastique humaine qui sonde 84 protéines différentes de liaison à l'ADN et 11 modifications chimiques des histones. Les protéines histones aident à organiser le génome en agissant comme des bobines autour desquelles l'ADN s'enroule.
En utilisant les données de quelques-uns des chromosomes, ils ont entraîné un réseau neuronal personnalisé appelé MEGABASE (Maximum Entropy Genomic Annotation from Biomarkers Associated with Structural Ensembles) pour produire une séquence de types de chromatine. Cela a révélé comment les marques épigénétiques étaient liées aux compartiments, ils ont dit. Une fois formé, ils ont validé le modèle MEGABASE en lui fournissant les données des chromosomes restants. Cela a produit un nouvel ensemble de types structurels pour analyse par le programme MiChroM de l'équipe Rice, un cousin de l'algorithme de paysage énergétique AWSEM du laboratoire qui prédit les structures des protéines. L'algorithme MiChroM a prédit les structures 3-D des chromosomes.
"Nos résultats soutiennent l'idée que la compartimentation dans les chromosomes résulte de la séparation de phases de différents types de chromatine dans le noyau, comme la séparation de l'huile et de l'eau, " dit Cheng.
Lorsque les chercheurs ont réduit l'ensemble de données d'origine à seulement 11 marques d'histones et ont réexécuté les calculs, les résultats n'étaient que légèrement différents. Finalement, ils ont déterminé que les données histones suffisent à elles seules à prédire la forme d'un chromosome. "Il y a un code bien défini qui relie les marques d'histone à la structure, " dit Di Pierro. " C'est bien conservé, il est donc probable qu'il ait une fonction."
Pour valider leur théorie, les chercheurs ont comparé leurs résultats avec les cartes de contact des cellules lymphoblastes générées par Hi-C. Cette technique expérimentale, qui utilise le séquençage à haut débit pour identifier les modèles de repliement dans l'ADN, a été développé par le co-auteur Erez Lieberman Aiden, directeur du Baylor's Center for Genome Architecture et chercheur principal au CTBP.
"Cet article dit que nous pouvons prendre des informations unidimensionnelles sur les histones et les utiliser avec nos outils de Big Data pour prédire la structure tridimensionnelle, " a déclaré Wolyne.
Leur succès rapproche l'équipe de l'objectif ultime d'une théorie qui prédit l'architecture d'un génome entier. Cependant, il reste un problème de poule ou d'œuf :la chromatine se replie-t-elle à cause des marqueurs, ou les marqueurs apparaissent à cause du pliage ?
"Cela fait partie de notre fascination pour la façon dont la vie fonctionne, " a déclaré Di Pierro. "C'est un beau problème."














