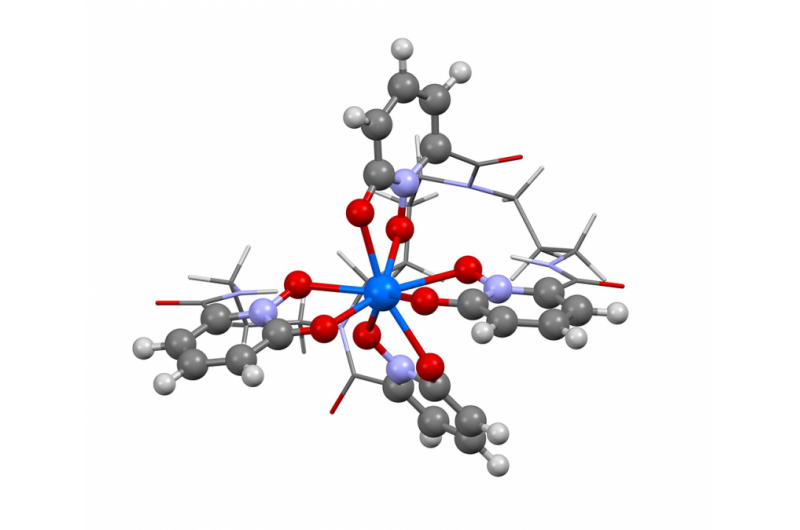
Cette image montre la structure du berkelium à l'état d'oxydation +IV. Les chercheurs ont utilisé le nouvel algorithme de Berkeley Lab pour calculer le spectre d'absorption et confirmer ce que plusieurs résultats expérimentaux laissaient entendre - que l'élément berkelium se brise avec ses pairs éléments lourds en prenant une charge positive supplémentaire lorsqu'il est lié à une molécule organique synthétique. Cette propriété pourrait aider les scientifiques à développer de meilleures méthodes de manipulation et de purification des matières nucléaires. Crédit :Bert de Jong, Laboratoire de Berkeley
Les objets qui brillent dans le noir semblent magiques quand on est enfant :ils peuvent illuminer une pièce sombre sans avoir besoin d'électricité, piles ou une ampoule. Puis, à un moment donné, vous apprenez la science derrière ce phénomène. Les composés chimiques appelés chromophores deviennent énergisés, ou excité, quand ils absorbent la lumière visible. En revenant à leur état normal, l'énergie stockée est libérée sous forme de lumière, que nous percevons comme une lueur. En science des matériaux, les chercheurs s'appuient sur un phénomène similaire pour étudier les structures des matériaux qui seront éventuellement utilisés en catalyse chimique, piles, applications solaires et plus.
Lorsqu'une molécule absorbe un photon, la particule fondamentale de la lumière, les électrons du système moléculaire passent d'un état de basse énergie (fond) à un état d'énergie plus élevée (excité). Ces réponses résonnent à des fréquences lumineuses spécifiques, laissant des "empreintes spectrales" qui éclairent les structures atomiques et électroniques du système étudié.
Dans les expériences, les "empreintes spectrales" ou spectre d'absorption, sont mesurées avec des installations de pointe telles que la source lumineuse avancée (ALS) du Lawrence Berkeley National Laboratory (Berkeley Lab) du département américain de l'Énergie. Dans les simulations informatiques, ces mesures sont généralement capturées avec une méthode de mécanique quantique appelée théorie fonctionnelle de la densité dépendante du temps (TDDFT). Les modèles informatiques sont essentiels pour aider les chercheurs à tirer le meilleur parti de leurs expériences en prédisant et en validant les résultats.
Pourtant, malgré son utilité, il y a des moments où TDDFT ne peut pas être utilisé pour calculer le spectre d'absorption d'un système car cela nécessiterait trop de temps et de ressources informatiques. C'est là qu'un nouveau "raccourci" mathématique développé par des chercheurs de la division de recherche informatique (CRD) de Berkeley Lab s'avère utile. Leur algorithme accélère les calculs d'absorption d'un facteur cinq, Ainsi, les simulations qui prenaient 10 à 15 heures à calculer peuvent désormais être effectuées en environ 2,5 heures.
Un article décrivant cette méthode a été publié dans le Journal of Chemical Theory and Computation (JCTC). Et la nouvelle approche de calcul du spectre d'absorption sera intégrée dans une prochaine version de la suite logicielle de chimie computationnelle largement utilisée NWchem plus tard cette année.
De nouveaux algorithmes permettent de réaliser des économies de calcul
Pour étudier la structure chimique de nouvelles molécules et matériaux, les scientifiques sondent généralement le système avec un stimulus externe, généralement un laser, puis recherchent de petits changements électroniques. Mathématiquement, ce changement électronique peut être exprimé comme un problème aux valeurs propres. En résolvant ce problème aux valeurs propres, les chercheurs peuvent obtenir une bonne approximation du spectre d'absorption, qui à son tour révèle les fréquences de résonance du système étudié. Pendant ce temps, le vecteur propre correspondant est utilisé pour calculer l'intensité avec laquelle le système a répondu au stimulus. C'est essentiellement le principe de l'approche TDDFT, qui a été implémenté dans plusieurs progiciels de chimie quantique, y compris la suite logicielle open source NWchem.
Bien que cette approche ait fait ses preuves, il a des limites pour les grands systèmes. Plus la gamme d'énergie des réponses électroniques qu'un chercheur essaie de capturer dans un système est large, plus il faut calculer de valeurs propres et de vecteurs propres, ce qui signifie également que davantage de ressources informatiques sont nécessaires. Finalement, le spectre d'absorption d'un système moléculaire avec plus de 100 atomes devient prohibitif à calculer avec cette méthode.
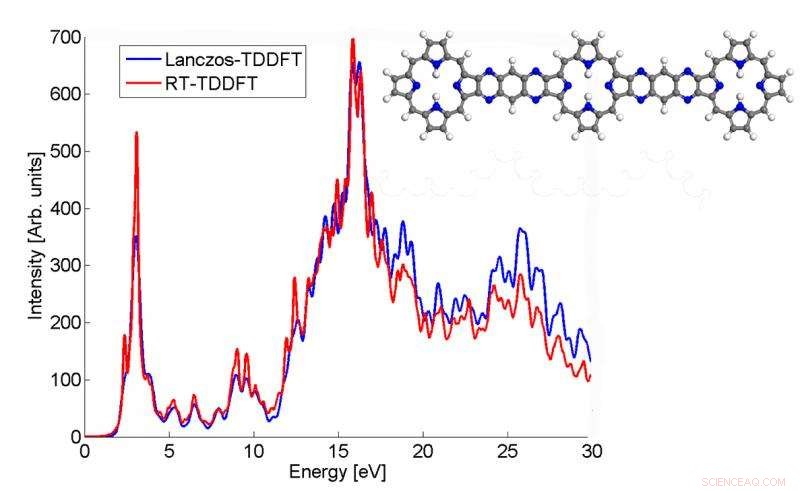
Ce graphique montre comment le spectre d'absorption d'une molécule p3b2 calculé par l'algorithme de Lanczos correspond au résultat TDDFT en temps réel. Crédit :Chao Yang, Laboratoire de Berkeley
Pour surmonter ces limites, les mathématiciens du CRD ont développé une technique pour calculer le spectre d'absorption directement sans calculer explicitement les valeurs propres de la matrice.
"Traditionnellement, les chercheurs ont dû calculer les valeurs propres et vecteurs propres de très grandes matrices afin de générer le spectre d'absorption, mais nous avons réalisé que vous n'avez pas à calculer chaque valeur propre pour obtenir une vue précise du spectre d'absorption, " dit Chao Yang, un mathématicien du CRD qui a dirigé le développement de la nouvelle approche.
En reformulant le problème comme une approximation de fonction matricielle, en utilisant une transformation spéciale et en tirant parti de la symétrie sous-jacente par rapport à une métrique non euclidienne, Yang et ses collègues ont pu appliquer l'algorithme de Lanczos et une méthode polynomiale du noyau (KPM) pour approximer le spectre d'absorption de plusieurs molécules. Ces deux algorithmes nécessitent une mémoire relativement faible par rapport aux alternatives non symétriques, qui est la clé des économies de calcul.
Parce que cette méthode nécessite moins de puissance de calcul pour obtenir un résultat, les chercheurs peuvent aussi facilement calculer le spectre d'absorption pour des systèmes moléculaires de plusieurs centaines d'atomes.
"Cette méthode est une avancée significative car elle nous permet de modéliser le spectre d'absorption de systèmes moléculaires de centaines d'atomes à un coût de calcul inférieur." dit Niranjan Govind, un chimiste informatique au Pacific Northwest National Laboratory qui a collaboré avec l'équipe de Berkeley Lab sur le développement de la méthode dans le programme de chimie informatique NWchem.
Récemment, les scientifiques de Berkeley Lab ont utilisé cette méthode pour calculer le spectre d'absorption et confirmer ce que plusieurs résultats expérimentaux ont laissé entendre - que l'élément berkelium se brise avec ses pairs d'éléments lourds en prenant une charge positive supplémentaire lorsqu'il est lié à une molécule organique synthétique. Cette propriété pourrait aider les scientifiques à développer de meilleures méthodes de manipulation et de purification des matières nucléaires. Un article soulignant ce résultat est paru le 10 avril dans la revue Chimie de la nature .
"Les résultats expérimentaux faisaient allusion à ce comportement inhabituel du berkelium, mais il n'y avait pas assez de preuves expérimentales pour dire oui, 100 pourcent, c'est ce que nous voyons, " déclare le co-auteur de l'étude Wibe Albert de Jong, un scientifique du CRD. "Pour être sûr à 100 pour cent, nous avons fait de grandes simulations informatiques et les avons comparées aux données expérimentales et déterminé qu'elles étaient, En effet, voir le berkélium dans un état d'oxydation inhabituel."
Ce nouvel algorithme a été développé dans le cadre d'un projet SciDAC (Scientific Discovery through Advanced Computing) soutenu par le DOE Office of Science et axé sur l'avancement des logiciels et des algorithmes pour les réactions photochimiques. Les projets SciDAC rassemblent généralement une équipe interdisciplinaire de chercheurs pour développer de nouvelles méthodes de calcul pour résoudre certains des problèmes scientifiques les plus difficiles.
« La nature interdisciplinaire de SciDAC est un moyen très efficace de faciliter les avancées scientifiques, comme chaque membre de l'équipe apporte une perspective différente à la résolution de problèmes, " dit Yang. " Dans cet environnement dynamique, mathématiciens, comme moi, faire équipe avec des scientifiques du domaine pour identifier les goulots d'étranglement de calcul, puis nous utilisons des techniques mathématiques de pointe pour relever et surmonter ces défis. »