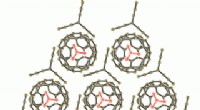
GABA
L'examen de la disposition tridimensionnelle précise des atomes au sein d'une protéine nous aide à comprendre comment elle peut remplir ses fonctions. Bien que la cryo-microscopie électronique (cryo-EM) se soit développée rapidement en tant que technique importante de biologie structurale ces dernières années, La cristallographie aux rayons X avait été la seule technique capable de visualiser des atomes individuels. Les groupes de Radu Aricescu et Sjors Scheres au Laboratoire de biologie moléculaire du MRC, en collaboration avec des scientifiques de Thermo Fisher Scientific et d'ailleurs, ont maintenant été capables de résoudre des atomes de protéines individuels pour la première fois dans une image cryo-EM tridimensionnelle.
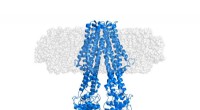
Cette collaboration a débuté début 2019 lorsque Radu et Abhay Kotecha, chercheur chez Thermo Fisher Scientific, voulait tester un nouveau matériel cryo-EM sur un petit échantillon de protéine membranaire. récepteurs GABAA, au centre des recherches de Radu depuis plus d'une décennie, ont été choisis parce que la résolution la plus élevée possible en utilisant la meilleure technologie disponible semblait avoir atteint une limite à environ 2,5 Ångströms (Å), mais une résolution plus élevée était clairement nécessaire pour une meilleure conception des médicaments.
Qu'est-ce que la résolution atomique ?
La résolution est généralement indiquée en Ångströms, une unité de longueur qui est un dix milliardième de mètre ou 0,1 nanomètre, et se réfère à la plus petite distance entre laquelle deux objets peuvent être considérés comme séparés.
La longueur d'une liaison carbone-carbone typique est de 1,5; d'autres liaisons dans les protéines sont un peu plus courtes. Ainsi, lorsque la résolution descend à 1,2 Å, il devient possible de voir des atomes individuels au sein d'une protéine, obtenir une véritable résolution atomique.
Tout en testant de nouveaux développements matériels qui comprenaient une source d'électrons à canon à émission de champ froid, un nouveau filtre énergétique, et un nouvel appareil photo, l'équipe a également dû développer de nouvelles stratégies de traitement. Algorithmes de correction des aberrations optiques précédemment développés par Jasenko Zivanov dans le groupe de Sjors, ainsi qu'un algorithme proposé par Chris Russo et Richard Henderson, joué un rôle crucial en extrayant le plus d'informations des images.
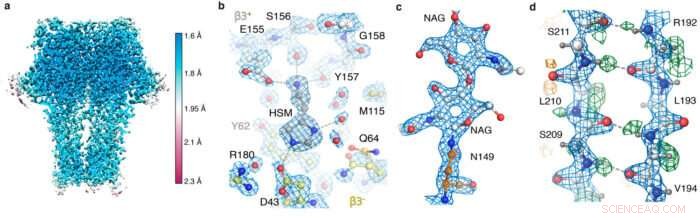
Après avoir reçu des images collectées sur le nouveau matériel de microscope par Abhay Kotecha chez Thermo Fisher Scientific à Eindhoven, Pays-Bas, Takanori Nakane, un post-doctorant dans le groupe Sjors, développé un workflow optimal dans RELION et Andrija Sente, avec d'autres membres du groupe de Radu, utilisé ce flux de travail pour traiter les images des récepteurs GABAA, tout en restituant les résultats pour optimiser rapidement les réglages du microscope. Un nouveau, Le système de stockage de données haute capacité développé par Jake Grimmett et Toby Darling dans l'équipe de calcul scientifique du LMB a offert un support crucial pour gérer les quelque cent téraoctets de données générées. Cet effort soutenu de l'équipe a conduit à une structure de récepteur GABAA d'une résolution sans précédent de 1,7 .
Il s'agit de la meilleure résolution rapportée obtenue en utilisant cryo-EM pour tout échantillon de protéine autre que la protéine apoferritine. L'apoferritine est couramment utilisée comme référence pour la cryo-EM, parce que sa stabilité moléculaire et sa symétrie 24 fois permettent des reconstructions à haute résolution à partir de relativement peu de particules.
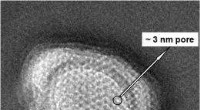
En utilisant les nouvelles stratégies de matériel et de traitement, l'équipe a pu obtenir une structure d'apoferritine de résolution 1,22 , battant le précédent record de 1,53 pour être la structure cryo-EM monoparticulaire la plus haute résolution jamais obtenue. Le plus impressionnant, cette résolution a permis de visualiser des atomes d'hydrogène individuels, même sur les molécules d'eau à l'intérieur de la structure protéique. La visualisation des réseaux de liaisons hydrogène à l'intérieur des structures protéiques et dans les poches de liaison aux médicaments permet aux chercheurs de mieux comprendre leur fonctionnement.
Ce travail représente la rupture d'une barrière clé pour la cryo-EM en tant que technique de biologie structurale et la nouvelle technologie, collecte de données, et les stratégies de traitement augmenteront le nombre de protéines dont les structures peuvent être résolues à haute résolution. Ces reconstructions à plus haute résolution permettront de mieux comprendre le fonctionnement des protéines et faciliteront la conception de médicaments plus spécifiques qui pourraient avoir un impact sur les traitements d'un large éventail de maladies.