Une analyse à haut débit des compositions possibles pour une nouvelle classe de matériaux connue sous le nom de MXenes donne aux chercheurs une direction inestimable pour choisir le meilleur candidat parmi les millions de recettes de matériaux possibles. L'étude de simulation par des chercheurs de l'A*STAR Institute of High Performance Computing est une avancée significative dans le domaine des MXenes, qui ont un potentiel passionnant dans les applications de stockage d'énergie de nouvelle génération.
Les matériaux bidimensionnels (2-D) sont une classe de matériaux relativement nouvelle qui présentent un large éventail de propriétés inhabituelles associées à leur capacité à contraindre le mouvement des électrons et de l'énergie dans un plan 2-D. Les alliages MXene sont une classe de matériaux 2-D découverte très récemment, qui pourrait consister en n'importe lequel des millions d'arrangements possibles de métaux de transition (comme le molybdène ou le titane), carbone et azote. Ces caractéristiques se reflètent dans le nom « MXene » - le « M » représente les atomes de métal, le 'X' désigne le carbone et l'azote, tandis que le suffixe 'ene' signale la structure atomique 2-D des matériaux.
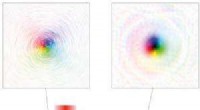
"Comme les MXenes sont nouveaux, il y a encore beaucoup à apprendre sur leur structure et leurs propriétés, " dit Teck Leong Tan de A*STAR. "Comme les alliages MXene sont formés en mélangeant différents types d'éléments de transition à différentes compositions, les possibilités d'alliage dans MXenes sont énormes. Nous avons donc développé une méthode de calcul à haut débit pour prédire les structures probables et les phases stables de différents alliages de MXene dans toutes les plages de composition et de températures. »
Bien qu'il existe de nombreuses compositions possibles d'alliage MXene, la plupart ne seront pas stables. Le défi auquel sont confrontés les scientifiques des matériaux a été de savoir comment balayer efficacement le grand nombre de configurations d'alliages pour identifier celles ayant l'énergie de formation la plus faible et donc la stabilité la plus élevée. Les approches conventionnelles de calcul des « premiers principes » sont trop gourmandes en calculs pour qu'une telle analyse soit réalisable.
"Notre approche utilise ce qu'on appelle une méthode d'expansion de cluster pour "apprendre" les interactions efficaces entre les atomes, permettant ainsi une évaluation rapide des énergies de formation de millions de structures en alliage MXene, " dit Tan.
Le scan, menée en collaboration avec l'Université Drexel aux États-Unis, a révélé que des MXènes à base de molybdène mélangés à du vanadium, tantale, niobium ou titane, semblent former les structures ordonnées les plus stables. Le titane a cependant tendance à former des structures ordonnées «asymétriques» stables qui n'étaient auparavant pas considérées comme viables.
"Notre scan nous permet de prédire les structures des alliages MXene qui doivent encore être fabriqués et d'estimer la probabilité de leur fabrication d'un point de vue thermodynamique. Et pour les alliages MXene connus, nos structures prédites sont cohérentes avec les résultats expérimentaux."