Les membres de CAMERA (de gauche à droite) Peter Zwart, Jeff Donatelli et Kanupriya Pande, co-auteurs d'un article décrivant comment l'algorithme M-TIP du groupe a déterminé les structures virales 3D à partir de données de diffraction de particules uniques. Donatelli détient un modèle imprimé en 3D de l'un des virus reconstruits par M-TIP. Crédit :Marilyn Chung, Laboratoire de Berkeley
Au sein d'une équipe de recherche internationale, Jeff Donatelli, Peter Zwart et Kanupriya Pande du Center for Advanced Mathematics for Energy Research Applications (CAMERA) du Lawrence Berkeley National Laboratory (Berkeley Lab) ont contribué aux algorithmes clés qui ont aidé à atteindre un objectif proposé pour la première fois il y a plus de 40 ans - en utilisant des corrélations angulaires de rayons X des instantanés de molécules non cristallines pour déterminer la structure 3D d'objets biologiques importants. Cette technique a le potentiel de permettre aux scientifiques de faire la lumière sur la structure et la dynamique biologiques qui étaient auparavant impossibles à observer avec les méthodes de rayons X traditionnelles.
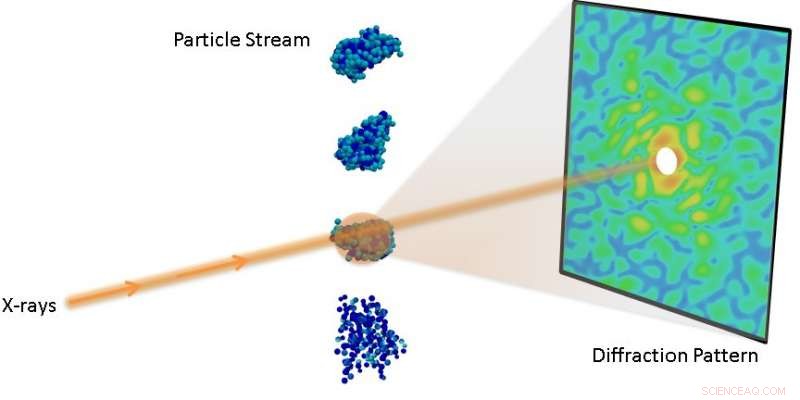
Cette percée est le résultat d'une expérience de diffraction à particule unique menée à la source de lumière cohérente Linac (LCLS) du ministère de l'Énergie (DOE) par l'Initiative à particule unique organisée par le SLAC National Accelerator Laboratory. Dans le cadre de cette initiative, l'équipe CAMERA a combiné ses efforts avec Ruslan Kurta, un physicien de l'installation européenne XFEL (laser à électrons libres à rayons X) en Allemagne, pour analyser les corrélations angulaires à partir des données expérimentales et utiliser l'algorithme de phasage itératif à plusieurs niveaux (M-TIP) de CAMERA pour effectuer les premières reconstructions de virus 3D réussies à partir de corrélations expérimentales. Les résultats ont été décrits dans un article publié le 12 octobre dans Lettres d'examen physique .
« Depuis 40 ans, cela a été considéré comme un problème qui ne pouvait pas être résolu, " dit Peter Zwart, co-auteur de l'article et bioscientifique physique membre de CAMERA basé à la division de biophysique moléculaire et d'imagerie intégrée du Berkeley Lab. "Mais il s'avère que les outils mathématiques que nous avons développés sont capables d'exploiter des informations supplémentaires cachées dans le problème qui avaient été précédemment négligées. Il est gratifiant de voir notre approche théorique conduire à un outil pratique."
Nouvelles opportunités de recherche rendues possibles par les XFEL
Pendant une bonne partie du siècle dernier, la technique de prédilection pour déterminer la structure moléculaire à haute résolution a été la cristallographie aux rayons X, où l'échantillon d'intérêt est disposé dans un grand réseau périodique et exposé aux rayons X qui se dispersent et forment des motifs de diffraction qui sont collectés sur un détecteur. Même si la cristallographie a réussi à déterminer de nombreuses structures à haute résolution, il est difficile d'utiliser cette technique pour étudier des structures qui ne sont pas sensibles à la cristallisation ou à des changements structurels qui ne se produisent pas naturellement dans un cristal.
La création des installations XFEL, y compris la source de lumière cohérente Linac (LCLS) et le X-FEL européen, ont créé des opportunités pour mener de nouvelles expériences qui peuvent surmonter les limites de la cristallographie traditionnelle. En particulier, Les faisceaux XFEL sont de plusieurs ordres de grandeur plus brillants et ont des durées d'impulsion beaucoup plus courtes que les sources lumineuses à rayons X traditionnelles, qui leur permettent de collecter un signal de diffraction mesurable à partir d'échantillons non cristallisés plus petits et d'étudier également la dynamique rapide. La diffraction de particules uniques est l'une de ces techniques expérimentales émergentes activées par XFELS, où l'on collecte des images de diffraction de molécules uniques au lieu de cristaux. Ces techniques à particule unique peuvent être utilisées pour étudier la structure et la dynamique moléculaires qui ont été difficiles à étudier avec les techniques d'imagerie traditionnelles.
Surmonter les limites de l'imagerie à particule unique via des corrélations angulaires
Un défi majeur de l'imagerie monoparticulaire est celui de la détermination de l'orientation. "Dans une expérience à une seule particule, vous n'avez pas de contrôle sur la rotation des particules car elles sont frappées par le faisceau de rayons X, ainsi chaque instantané d'un hit réussi contiendra des informations sur l'échantillon d'une orientation différente, " a déclaré le co-auteur Jeff Donatelli, un mathématicien appliqué dans CAMERA qui a développé de nombreux algorithmes dans le nouveau cadre. « La plupart des approches de l'analyse de particules uniques ont jusqu'à présent été basées sur la détermination de ces orientations de particules à partir des images ; cependant, la meilleure résolution pouvant être obtenue à partir de ces analyses est limitée par la précision avec laquelle ces orientations peuvent être déterminées à partir de données bruitées."
Au lieu d'essayer de déterminer directement ces orientations, l'équipe a adopté une approche différente basée sur l'idée initialement proposée dans les années 1970 par Zvi Kam. "Plutôt que d'examiner les intensités de données individuelles pour tenter de trouver l'orientation correcte pour chaque image mesurée, nous éliminons complètement cette étape en utilisant des fonctions dites de corrélation croisée, " dit Kurta.
Cette approche, connue sous le nom de diffusion des rayons X par fluctuation, est basé sur l'analyse des corrélations angulaires de l'ultracourt, intenses impulsions de rayons X diffusées à partir de biomolécules non cristallines. "La beauté de l'utilisation des données de corrélation est qu'elles contiennent une empreinte complète de la structure 3D d'un objet qui étend les approches traditionnelles de diffusion de solution, " a déclaré Zwart.
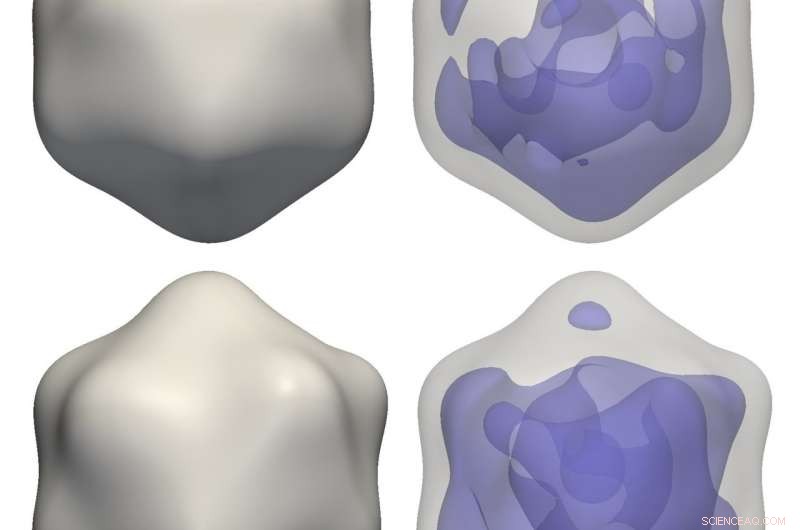
Virus reconstruits :Reconstructions d'un virus nain du riz (en haut) et d'un bactériophage PR772 (en bas) à partir de données de corrélation expérimentales utilisant M-TIP. Les images de droite montrent des asymétries dans le matériel génétique interne pour chaque reconstruction virale. Crédit :Jeff Donatelli, Laboratoire de Berkeley
Reconstruire la structure 3D à partir de corrélations avec l'algorithme M-TIP de CAMERA
La percée de l'équipe dans la reconstruction de la structure 3D à partir des données de corrélation a été rendue possible par l'algorithme de phasage itératif à plusieurs niveaux (M-TIP) développé par CAMERA. « L'un des principaux avantages de M-TIP est sa capacité à résoudre la structure directement à partir des données de corrélation sans avoir à s'appuyer sur des contraintes de symétrie, et, plus important, sans qu'il soit nécessaire de résoudre le problème de détermination de l'orientation, " a déclaré Donatelli.
Donatelli, Le directeur de CAMERA James Sethian et Zwart ont développé leur cadre M-TIP en développant une généralisation mathématique d'une classe d'algorithmes connus sous le nom de techniques de phasage itératif, qui sont utilisés pour déterminer la structure dans un problème plus simple, connu sous le nom de récupération de phase. Un article décrivant le cadre original du M-TIP a été publié en août 2015 dans le Actes de l'Académie nationale des sciences .
"Les analyses de corrélation avancées en combinaison avec les reconstructions ab-initio par M-TIP définissent clairement une voie efficace pour l'analyse structurelle d'objets nanométriques aux XFEL, " a déclaré Zwart.
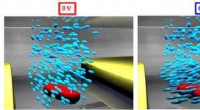
Montage expérimental pour une expérience de diffraction à une seule particule. Crédit :Lawrence Berkeley National Laboratory
Orientations futures de l'analyse de corrélation et du M-TIP
L'équipe note que les méthodes utilisées dans cette analyse peuvent également être appliquées pour analyser les données de diffraction lorsqu'il y a plus d'une particule par tir.
"La plupart des algorithmes d'imagerie à particule unique ne peuvent gérer qu'une molécule à la fois, limitant ainsi le signal et la résolution. Notre approche, d'autre part, est évolutif de sorte que nous devrions également pouvoir mesurer plus d'une particule à la fois, " a déclaré Kurta. L'imagerie avec plus d'une particule par tir permettra aux scientifiques d'atteindre des taux de réussite beaucoup plus élevés, puisqu'il est plus facile d'utiliser un faisceau large et de frapper plusieurs particules à la fois, et évitera également d'avoir à séparer les coups à particule unique des coups à particules multiples et les coups à blanc, ce qui est une autre exigence difficile dans l'imagerie traditionnelle à particule unique.
Dans le cadre de la suite d'outils de calcul de CAMERA, ils ont également développé une version différente de M-TIP qui résout le problème d'orientation et peut classer les images en états conformationnels, et par conséquent peut être utilisé pour étudier de petites différences biologiques dans l'échantillon mesuré. Cette version alternative de M-TIP a été décrite dans un article publié le 26 juin 2017 dans le Actes de l'Académie nationale des sciences et fait partie d'une nouvelle initiative de collaboration entre le SLAC National Accelerator Laboratory, CAMERA, the National Energy Research Scientific Computing Center (NERSC) and Los Alamos National Laboratory as part of DOE's Exascale Computing Project (ECP).