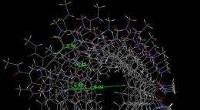
Les calculs montrent que les nanoparticules à noyau de platine (gris)-coquille de palladium (vert) sont particulièrement stables. Les atomes d'hydrogène (rouge) qui s'adsorbent à la surface des particules sont convertis catalytiquement en hydrogène gazeux. Crédit : 2012 A*STAR Institute of High Performance Computing
Les nanoparticules peuvent être de puissants catalyseurs. Nano-alliages bimétalliques de platine et de palladium, par exemple, peut aider à produire de l'hydrogène comme combustible en favorisant la décomposition électrochimique de l'eau. Identifier le nano-alliage le plus actif pour une telle tâche, cependant, reste un défi; les performances catalytiques sont directement liées à la structure des particules, et les expériences pour établir l'arrangement atomique de ces petites particules sont difficiles à réaliser. Il est désormais possible de prédire des structures de nano-alliages stables à l'aide d'une approche informatique développée par Teck Leong Tan de l'A*STAR Institute of High Performance Computing et ses collègues. Leur technique peut également identifier des façons dont la structure atomique de la nanoparticule pourrait être ajustée pour améliorer les performances catalytiques.
Le défi du calcul de la structure et des propriétés des nano-alliages à partir des premiers principes est la puissance de traitement de calcul qu'il nécessite, dit Tan. Pour leur étude, lui et ses collègues ont considéré une particule de nano-alliage de 55 atomes, chaque site de la structure est rempli soit par un atome de palladium, soit par un atome de platine. "Il y a des millions de configurations d'alliages possibles, il serait donc difficile de faire une recherche directe en utilisant les calculs des premiers principes, " explique Tan.
Pour rendre le processus gérable, les chercheurs ont divisé conceptuellement la nanoparticule en petites sous-unités géométriques, ou des grappes. A partir des premiers calculs de principe sur un ensemble d'une centaine de structures d'alliages différentes, constitués chacun d'une trentaine de clusters, ils ont généré un modèle fiable du comportement de l'alliage en utilisant une approche appelée expansion de cluster. A partir de ce modèle, ils ont calculé les propriétés des nanoparticules entières. "Le modèle est utilisé pour rechercher rapidement dans l'immense espace de configuration des états de basse énergie, " dit Tan. Ces états de basse énergie représentent les configurations d'alliage stables qui devraient exister expérimentalement (voir image).
En utilisant leurs structures stables calculées, Tan et ses collègues ont ensuite prédit comment différentes conformations atomiques affectent les performances d'une particule en tant que catalyseur. Comme réaction modèle, les chercheurs ont examiné la réaction de dégagement d'hydrogène, la génération électrochimique d'hydrogène gazeux. Les résultats suggèrent que l'activité catalytique des particules augmentera à mesure que plus de palladium est ajouté, car cet alliage améliore la liaison de l'hydrogène à divers sites d'adsorption à la surface des nanoparticules, informations utiles pour guider la synthèse de nouveaux nanocatalyseurs.
L'approche devrait être largement applicable pour la recherche sur les nanoparticules, note Tan. "La méthode d'expansion de cluster peut généralement être appliquée à tous les systèmes d'alliages où les structures et les stabilités sont d'intérêt, " dit-il. Tan prévoit ensuite d'étudier l'impact des molécules adsorbées sur la surface d'un catalyseur. " La présence de molécules adsorbées conduit souvent à des changements dans les structures des alliages, altérant ainsi les performances catalytiques, " il dit.