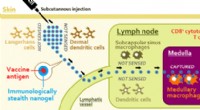
Pour étudier les nanostructures en environnement réel, Les scientifiques du Berkeley Lab ont combiné des approches théoriques et expérimentales pour entrevoir l'interaction d'une protéine avec des sels simples dans l'eau. Activé par un logiciel de simulation d'absorption des rayons X développé à la fonderie moléculaire de Berkeley Lab, ces découvertes jettent un nouvel éclairage sur l'impact des sels sur la structure des protéines au niveau atomique.
Techniques cristallographiques traditionnelles, comme la diffraction des rayons X, fournir un profil de matériaux ordonnés avec des structures statiques. Cependant, pour les systèmes dynamiques ou complexes dans lesquels la structure atomique évolue rapidement, des méthodes plus sophistiquées sont nécessaires. Maintenant, Les scientifiques du Berkeley Lab ont appliqué la spectroscopie d'absorption des rayons X pour étudier une protéine modèle, triglycine - une chaîne courte de trois molécules de l'acide aminé le plus simple, glycine. En simulant le spectre d'absorption des rayons X de cette molécule, l'équipe a montré comment sa chaîne se plie et se redresse en réponse aux ions en solution.
"Regarder une molécule en solution, c'est comme regarder une marionnette - vous pouvez la voir se plier en réponse à la création et à la rupture de liaisons hydrogène, " a déclaré David Prendergast, membre du personnel scientifique de l'installation de théorie des nanostructures de la fonderie moléculaire. « Une connaissance concrète de la façon dont les ions influencent ce comportement provient de l'utilisation de simulations de dynamique moléculaire, qui montrent des différences persistantes de structure sur des échelles de temps de la nanoseconde. À partir de ces données, nous pouvons générer des spectres d'absorption des rayons X qui peuvent ensuite être comparés aux résultats expérimentaux.
Dans une expérience d'absorption des rayons X spécialisée appelée structure fine d'absorption des rayons X près du bord (NEXAFS), les rayons X sont utilisés pour sonder la liaison chimique et l'environnement d'éléments spécifiques dans une molécule ou une nanostructure, tels que les atomes d'azote dans une molécule de triglycine. Couplé à une technologie de microjet liquide développée à Berkeley Labs, NEXAFS a déjà été utilisé pour examiner comment les protéines se dissolvent et cristallisent en présence de divers ions.
Le logiciel de Prendergast peut désormais simuler les données NEXAFS en faisant la moyenne d'une série d'instantanés pris à partir d'une simulation de dynamique moléculaire d'une molécule donnée. Ce logiciel est un outil essentiel pour interpréter les données NEXAFS à partir de données complexes, systèmes dynamiques, car les temps de sonde dans ces mesures sont trop lents - quelques secondes plutôt que des nanosecondes - pour révéler des différences structurelles à l'échelle nanométrique.
« Des études antérieures de notre groupe ont montré que le développement de la spectroscopie d'absorption des rayons X des microjets liquides fournit une nouvelle sonde atomique sensible des interactions entre les ions aqueux, mais c'est l'avènement de cette nouvelle théorie qui fournit la première interprétation fiable au niveau moléculaire de ces données, " a déclaré Richard Saykally, un chimiste du Berkeley Lab et professeur de chimie à l'Université de Californie à Berkeley. « Ici, nous voyons cette nouvelle combinaison de théorie et d'expérience appliquée à l'un des problèmes les plus importants de la chimie biophysique. »
Prendergast dit que sa technique de dynamique moléculaire peut être utilisée pour modéliser les spectres de rayons X d'un système biologique avec une structure connue pour déterminer ses interactions locales, ce qui l'amène à former une structure particulière, et pourquoi il prend une conformation particulière, le tout en simulant les spectres d'une série d'instantanés individuels et en les comparant aux résultats expérimentaux. Ces simulations sont gourmandes en calculs et reposent fortement sur l'infrastructure de supercalcul à grande échelle fournie par le Centre national de calcul scientifique de la recherche énergétique de Berkeley Lab (NERSC).
« Bien que ces effets soient une partie fondamentale de la nature, ils sont encore mal compris, " a déclaré Craig Schwartz, un chercheur travaillant avec Prendergast et Saykally, dont les travaux de fin d'études ont conduit à cette publication. « La sensibilité expérimentale de NEXAFS, couplé avec une percée dans la théorie, nous a donné un nouvel aperçu de la façon dont ces molécules interagissent.
Les chercheurs anticipent la demande d'autres groupes explorant les interactions eau (ou autres solvants), ainsi que les matériaux mous (tels que les polymères) et les matériaux inorganiques (oxydes et surfaces métalliques) qui sont directement pertinents pour les applications énergétiques en catalyse, technologie des batteries et photovoltaïque. En outre, à mesure que les sources laser à électrons libres à rayons X deviennent disponibles pour les scientifiques, un ensemble de données expérimentales plus riche sera disponible pour enrichir les découvertes théoriques.