Les scientifiques et ingénieurs en matériaux aimeraient savoir précisément comment les électrons interagissent et se déplacent dans les nouveaux matériaux et comment se comporteront les dispositifs fabriqués avec eux. Le courant électrique circulera-t-il facilement dans le matériau ? Existe-t-il une température à laquelle le matériau deviendra supraconducteur, permettant au courant de circuler sans source d'énergie ? Combien de temps l'état quantique du spin d'un électron sera-t-il préservé dans les nouveaux dispositifs électroniques et quantiques ?
Une communauté de physiciens des matériaux tente de répondre à ces questions en comprenant ce qui se passe à l'intérieur des matériaux, en calculant leur comportement jusqu'au niveau des interactions électroniques individuelles et des mouvements atomiques.
Aujourd'hui, une équipe de Caltech a fait une découverte clé qui permet de simplifier ces calculs, en les accélérant d'un facteur 50 ou plus tout en conservant leur précision. En conséquence, il est possible de calculer les interactions électroniques dans des matériaux et des dispositifs plus complexes ainsi que de développer de nouveaux calculs qui semblaient auparavant impossibles.
Dans un nouvel article publié dans la revue Physical Review X , Yao Luo de Caltech, étudiant diplômé en physique appliquée; son conseiller Marco Bernardi, professeur de physique appliquée, physique et science des matériaux; et ses collègues décrivent une nouvelle méthode basée sur les données qui a permis ces avancées. Leur approche simplifie les matrices informatiques denses utilisées pour représenter les interactions qui ont lieu dans un matériau entre les électrons et les vibrations atomiques (ou phonons, qui peuvent être considérés comme des unités individuelles d'énergie vibratoire).
Luo et Bernardi affirment que la nouvelle méthode leur permet d'utiliser seulement 1 à 2 % des données généralement utilisées pour résoudre de tels problèmes, accélérant considérablement les calculs et, ce faisant, révélant les interactions les plus importantes qui dictent les propriétés des matériaux. P>
"C'était très surprenant", déclare Bernardi. "Les interactions électron-phonon calculées avec les matrices compressées sont presque aussi précises que le calcul complet. Cela réduit considérablement le temps de calcul et l'utilisation de la mémoire, d'environ deux ordres de grandeur dans la plupart des cas. C'est aussi un exemple élégant du rasoir d'Occam, le idée de privilégier des modèles physiques simples avec un nombre minimal de paramètres."
Les chercheurs dans ce domaine suivent généralement l’une des deux approches suivantes pour comprendre les matériaux à ce niveau le plus fondamental. Une approche met l'accent sur la construction de modèles minimaux, réduisant ainsi la complexité du système, afin que les chercheurs puissent modifier une poignée de paramètres dans les calculs au stylo et sur papier pour obtenir une compréhension qualitative des matériaux.
L'autre commence par rien de plus que la structure d'un matériau et utilise des méthodes dites des « premiers principes » (calculs de mécanique quantique nécessitant de grands ordinateurs) pour étudier les propriétés des matériaux avec une précision quantitative.
Ce dernier ensemble de méthodes, sur lequel se concentre le groupe de Bernardi, utilise des matrices extrêmement grandes comportant des milliards d'entrées pour calculer les interactions électroniques qui contrôlent un large éventail de propriétés physiques. Cela se traduit par des milliers d’heures de temps de calcul pour chaque calcul. Le nouveau travail suggère une sorte de terrain d'entente entre les deux approches, dit Bernardi.
"Grâce à notre nouvelle méthode, vous pouvez tronquer la taille de ces matrices, extraire les informations clés et générer des modèles minimaux des interactions dans les matériaux."
L'approche de son groupe repose sur l'application d'une méthode appelée décomposition en valeurs singulières (SVD) aux interactions électron-phonon dans un matériau. La technique SVD est largement utilisée dans des domaines tels que la compression d’images et la science de l’information quantique. Ici, cela permet aux auteurs de séparer ou de démêler les composants électroniques et vibrationnels dans une matrice de milliers ou de millions d'interactions électron-phonon et d'attribuer un numéro à chaque interaction fondamentale.
Ces nombres réels positifs sont appelés valeurs singulières et classent les interactions fondamentales par ordre d’importance. Ensuite, le programme peut éliminer la quasi-totalité des interactions dans chaque matrice, sauf quelques pour cent, ne laissant que les principales valeurs singulières, un processus qui rend la détermination moins coûteuse d'un facteur proportionnel à la quantité de compression.
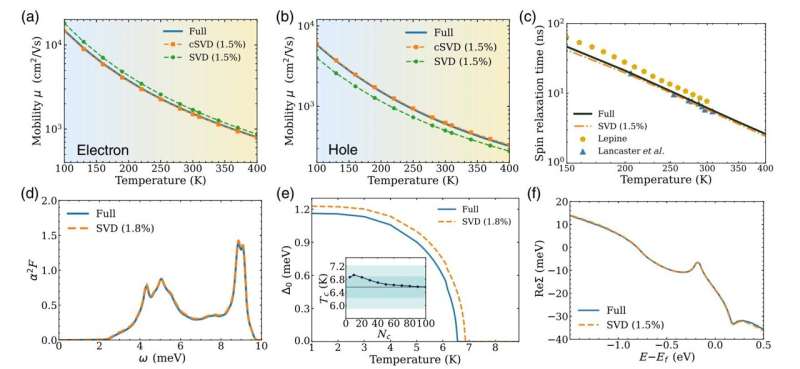
Ainsi, par exemple, si le programme ne conserve que 1 % des valeurs singulières, le calcul devient plus rapide d'un facteur 100. Les chercheurs ont constaté qu'en conservant seulement une petite fraction des valeurs singulières, généralement 1 à 2 %, le résultat approximatif conserve presque la même précision que le calcul complet.
"En utilisant SVD, vous pouvez réduire le nombre de valeurs singulières et capturer uniquement les principales caractéristiques des matrices représentant les interactions électroniques dans un matériau donné", explique Luo, auteur principal de l'article qui en est à sa troisième année dans le groupe de Bernardi. /P>
"Cela tronque la matrice d'origine, accélérant ainsi l'algorithme, et présente l'avantage supplémentaire de révéler quelles interactions dans le matériau sont dominantes."
Bernardi note que ce dernier avantage de la méthode SVD donne aux chercheurs une « intuition physique » sur les interactions électroniques dans un matériau, ce qui manquait dans les premiers calculs de principes dans le passé. Par exemple, dans un calcul impliquant du silicium, il est devenu clair que la valeur singulière dominante était associée à l'étirement et à la compression d'une liaison particulière.
"C'est quelque chose de simple, mais avant de faire le calcul, nous ne savions pas quelle était l'interaction la plus forte", explique Bernardi.
Dans cet article, les chercheurs montrent que la compression de matrices liées aux interactions électron-phonon à l'aide de la méthode SVD fournit des résultats précis pour diverses propriétés des matériaux que les chercheurs pourraient vouloir calculer, notamment le transport de charge, les temps de relaxation de spin et la température de transition des supraconducteurs. .
Bernardi et son équipe étendent les calculs basés sur SVD à une gamme plus large d'interactions dans les matériaux et développent des calculs avancés qui semblaient auparavant impossibles. L’équipe travaille également à ajouter la nouvelle méthode SVD à son code open source Perturbo, un progiciel qui aide les chercheurs à calculer la façon dont les électrons interagissent et se déplacent dans les matériaux. Bernardi affirme que cela permettra aux utilisateurs de la communauté scientifique de prédire beaucoup plus rapidement les propriétés des matériaux associées aux interactions électron-phonon.
L'article s'intitule « Compression basée sur les données des interactions électron-phonon ». Avec Luo et Bernardi, les co-auteurs de l'article comprennent l'étudiant diplômé Dhruv Desai (MS '22) ; Benjamin Chang (MS '20) et Jinsoo Park (Ph.D. '22), qui est maintenant chercheur postdoctoral à l'Université de Chicago.