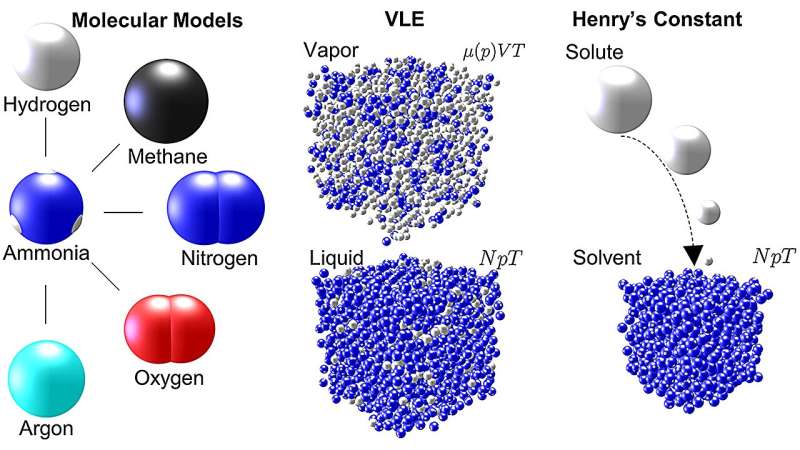
Ammoniac (NH3 ) est une molécule importante avec de nombreuses applications. Produit final du célèbre procédé Haber-Bosch, il est couramment synthétisé pour capturer l’azote destiné aux engrais et est utilisé pour la réfrigération, dans les produits de nettoyage et dans la production de produits pharmaceutiques. Récemment, cette modeste molécule a également suscité l'intérêt en tant que ressource potentielle pour relever l'un des défis les plus urgents d'aujourd'hui :le besoin de carburants renouvelables fiables et abondants.
L'ammoniac est stable et sûr à manipuler, est combustible et contient la plus grande fraction d'hydrogène de toutes les molécules, à l'exception de l'hydrogène pur lui-même. Ces facteurs promettent d’en faire une alternative réalisable aux vecteurs énergétiques basés sur le carbone qui sont à l’origine du changement climatique. Des recherches ont commencé pour explorer la manière dont l'ammoniac pourrait être utilisé pour alimenter directement des moteurs, des turbines à gaz et des piles à combustible à hydrogène, par exemple. On pense également que l'ammoniac pourrait être utilisé pour stocker de l'énergie lorsque d'autres énergies renouvelables comme l'énergie éolienne et solaire ne peuvent pas répondre à la demande.
On sait beaucoup de choses sur l’ammoniac, mais cet intérêt pour son utilisation comme combustible a lancé la recherche de nouvelles technologies liées à l’ammoniac. Cela a, à son tour, conduit à un besoin accru parmi les ingénieurs chimistes de données précises décrivant les propriétés thermodynamiques fondamentales de l'ammoniac. Ces propriétés incluent une grande variété de caractéristiques mesurables telles que les équilibres de phases, la densité ou la capacité thermique, par exemple, qui caractérisent les systèmes physiques et déterminent le fonctionnement des processus chimiques. Dans le cas de l’ammoniac, les ingénieurs souhaiteraient également mieux connaître la manière dont ces propriétés évoluent lors du mélange de l’ammoniac avec d’autres molécules. Ces connaissances pourraient les aider à optimiser les processus et les conditions de fonctionnement.
Le Dr Jadran Vrabec, actuellement directeur de l'Institut des sciences des procédés de l'Université technique de Berlin, a passé une grande partie de sa carrière à utiliser le calcul haute performance (HPC) pour étudier les propriétés thermodynamiques au niveau moléculaire. "Les propriétés thermodynamiques sont déterminées à 100 % par des interactions moléculaires", explique-t-il. "Et parce que ces interactions se produisent si rapidement et à si petite échelle, il n'est possible de les étudier qu'en effectuant de grandes simulations à l'aide de superordinateurs."
Dans un article récent publié dans le Journal of Chemical &Engineering Data , lui et le co-auteur Erich Mace de la TU Berlin rapportent les résultats de simulations axées sur les propriétés thermodynamiques de mélanges contenant de l'ammoniac. Réalisés à l'aide du supercalculateur Hawk du Centre de calcul haute performance de Stuttgart (HLRS), leurs résultats ajoutent des données précieuses qui pourraient soutenir le développement de nouvelles applications de l'ammoniac. Les résultats pourraient également aider à évaluer l'exactitude d'autres données existantes, garantissant ainsi que les ingénieurs disposent des meilleures informations disponibles pour travailler avec la substance.
Les simulations à grande échelle fournissent des informations uniques sur les propriétés thermodynamiques
Vrabec est un utilisateur de longue date des ressources de supercalcul HLRS pour la dynamique moléculaire et les simulations de Monte Carlo. Son approche s'appuie sur des concepts de thermodynamique qui ont été formulés pour la première fois par Ludwig Boltzmann au XIXe siècle, mais dont l'application n'est devenue pratique que dans les années 1950 avec l'arrivée des premiers ordinateurs. Depuis lors, le domaine a progressé parallèlement au développement de superordinateurs plus grands et plus rapides, au point que les simulations de Vrabec suivent désormais simultanément les mouvements individuels et les interactions de milliards, voire de milliards de molécules. À l'aide d'un logiciel développé par son laboratoire pour capturer sélectivement les données d'intérêt, il peut ensuite étudier les propriétés thermodynamiques des molécules.
Vrabec utilise deux codes de simulation appelés ms2 et ls1, qu'il a développés et optimisés au cours d'une longue et fructueuse collaboration avec Martin Bernreuther et Christoph Niethammer, membres du personnel du HLRS. En 2019, l’équipe a même établi un record mondial pour le plus grand système moléculaire jamais simulé à l’aide de méthodes de dynamique moléculaire. En utilisant ls1, ils ont efficacement adapté leur code à un système de 21 000 milliards d'atomes dans lequel chaque molécule individuelle et ses interactions avec d'autres molécules pouvaient être suivies.
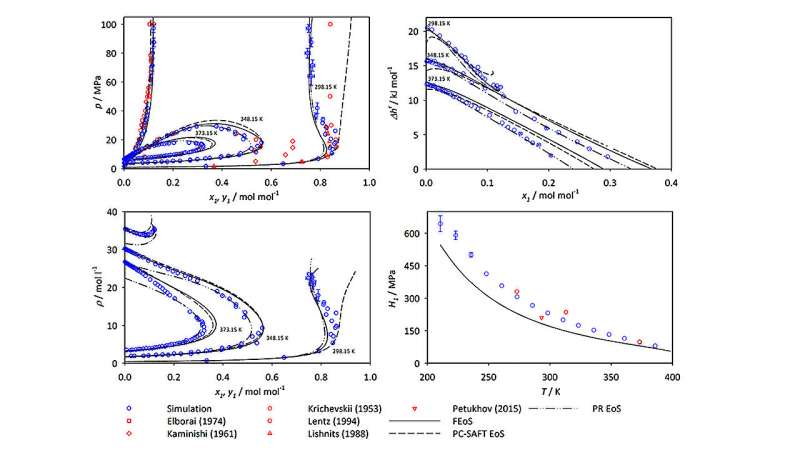
Dans leurs travaux récents sur l'ammoniac, Mace et Vrabec ont effectué des simulations de dynamique moléculaire et de Monte Carlo à l'aide de ms2 pour étudier cinq mélanges couramment utilisés impliquant de l'ammoniac dans les processus de génie chimique :argon-ammoniac, méthane-ammoniac, hydrogène-ammoniac, azote-ammoniac et oxygène. -ammoniac. Pour chaque mélange, les simulations ont généré des données décrivant l'équilibre vapeur-liquide (VLE) (une mesure de la distribution des molécules dans un système à travers les phases vapeur ou liquide) pour une large plage de températures et de pressions.
Dans leur article, Mace et Vrabec soulignent que les données VLE sont souvent utilisées pour développer des équations d'état pour les fluides industriels; c'est-à-dire que les données peuvent être utilisées pour prédire l'état de la matière dans différentes conditions physiques dues à des changements de température, de pression, de volume ou de composition. Ces informations sont essentielles pour déterminer les mélanges optimaux et les conditions de travail dans les applications industrielles.
Les simulations moléculaires de Vrabec sont particulièrement précieuses car elles peuvent être utilisées pour étudier une gamme d'échelles beaucoup plus large que ce qui est possible en utilisant des approches expérimentales.
"Dans nos simulations, nous avons fourni des mesures des propriétés thermodynamiques même jusqu'à des pressions de 50 mégapascals. C'est 500 fois notre pression de l'air ambiant", remarque Vrabec. "Bien que les données sur les mélanges d'ammoniac soient collectées depuis plus d'un siècle, la couverture des données est étonnamment étroite. La raison en est que les efforts nécessaires pour les mesurer expérimentalement sont d'une ampleur prohibitive. Cela nécessiterait un équipement spécial coûteux dont le fonctionnement serait dangereux. simulations informatiques, nous pouvons obtenir des résultats en toute sécurité et à relativement peu de frais. » Ses méthodes fournissent également un niveau de précision comparable à celui des approches expérimentales dans les domaines où des données expérimentales sont disponibles.
De meilleures données pour la recherche sur l'ammoniac
Lorsque Mace et Vrabec ont analysé leurs données de simulation, ils ont montré que bien que l'ammoniac soit un composant des cinq systèmes étudiés, les graphiques résultants des valeurs VLE semblent radicalement différents pour différents mélanges moléculaires. Selon Vrabec, "Le comportement de phase de différents mélanges est fortement déterminé par les interactions entre les molécules du système. Vous devez comprendre ces propriétés si vous souhaitez travailler avec des mélanges d'ammoniac."
L'article et ses données supplémentaires offrent plus de 400 nouveaux points de données pour chaque mélange étudié. Grâce à Hawk, ils ont pu produire les résultats de chaque mélange en quelques jours seulement de temps de calcul. Les résultats seront particulièrement précieux pour les conditions extrêmes et difficiles à étudier pour lesquelles peu de données sont disponibles, et pourraient aider les ingénieurs à identifier les points idéaux où les conditions seraient optimales pour un traitement efficace de l'ammoniac.
L'étude comprenait à la fois de nouvelles données de simulation et des données précédemment publiées dans la littérature scientifique, permettant à Mace et Vrabec de comparer leurs résultats avec d'autres ensembles de données existants sur les valeurs VLE. Dans la plupart des situations, leurs résultats correspondaient étroitement à ceux des études précédentes. Dans certains cas, cependant, ils ont identifié des divergences significatives entre leurs résultats et les mesures et prédictions générées expérimentalement par d’autres groupes de recherche. Les auteurs attribuent ces écarts aux limitations ou aux inexactitudes des méthodes expérimentales correspondantes. Ils suggèrent également que des sources de données expérimentales spécifiques doivent être utilisées avec prudence dans les futures recherches ou applications en génie chimique.
Vrabec dit que dans ses travaux récents, il s'est concentré principalement sur la simulation des propriétés thermodynamiques des systèmes moléculaires, généralement à l'échelle submicrométrique. Malgré les nombreux ordres de grandeur qui se situent entre cette échelle et le niveau des processus observables, des méthodes précises existent pour traduire ces informations au niveau moléculaire en prédictions utiles du monde réel.
Cependant, à mesure que les superordinateurs grandissent, il prévoit qu'il pourrait également devenir possible de simuler non seulement des propriétés, mais également des processus thermodynamiques en utilisant des conditions aux limites proches des applications du monde réel. Des performances HPC accrues pourraient produire des résultats plus précis sur les phénomènes dynamiques avec un meilleur rapport signal/bruit.
En attendant, les résultats de son équipe démontrent la valeur de la dynamique moléculaire et de la simulation Monte Carlo utilisant le calcul haute performance, et fourniront une nouvelle compréhension du comportement des phases que les ingénieurs pourront utiliser pour développer de nouvelles technologies basées sur l'ammoniac.
Plus d'informations : Erich J. Mace et al, Equilibres de phase fluide à haute pression et constantes de Henry des gaz supercritiques dans l'ammoniac, Journal of Chemical &Engineering Data (2023). DOI :10.1021/acs.jced.3c00327
Fourni par le Gauss Center for Supercomputing