Une méthode informatique permettant de trouver des états de transition dans des réactions chimiques, réduisant considérablement les coûts de calcul avec une grande fiabilité, a été conçue. Par rapport à la méthode existante la plus largement utilisée, la présente méthode réduit le coût de calcul total d'environ 50 à 70 %.
Le développement, disponible sur GitHub, est sur le point d'accélérer les progrès de la science des matériaux, rendant l'exploration des réactions chimiques plus accessible et plus efficace. Cela pourrait conduire à des découvertes scientifiques et à des innovations technologiques plus rapides.
Dans les réactions chimiques, les substances passent d’un état énergétiquement stable à un autre, en passant par un état de transition instable. Ce processus s’apparente à la recherche de l’itinéraire le plus bas au-dessus d’une montagne lors d’une traversée d’un côté à l’autre. Comprendre l'état de transition – le sommet de ce chemin de montagne métaphorique – est crucial pour une compréhension approfondie des mécanismes de réaction.
Cependant, en raison de la nature transitoire et instable de ces états, leur observation et leur identification expérimentales sont difficiles, nécessitant souvent une exploration informatique.
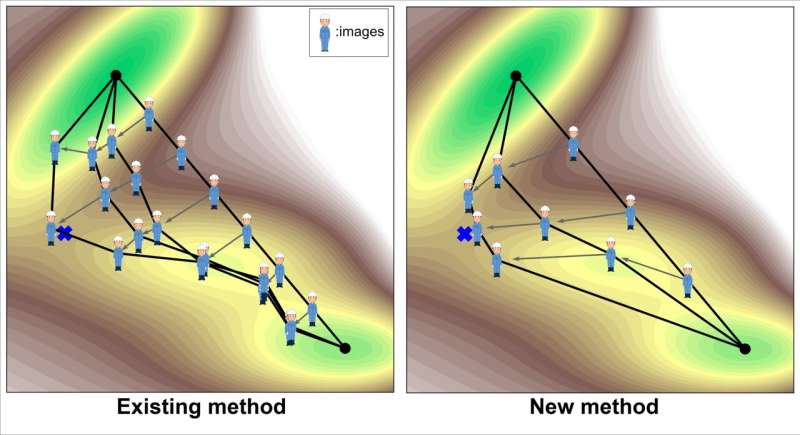
Cette étude se concentre sur les méthodes informatiques permettant de trouver un état de transition entre un réactif connu et un produit. Ce type de recherche d'état de transition optimise le chemin reliant le produit et le réactif afin qu'il passe par l'état de transition. Étant donné que le chemin est généralement représenté par plusieurs points sur le chemin (souvent appelés images), le chemin est en fait optimisé en mettant à jour progressivement les images.
La méthode la plus couramment utilisée aujourd’hui est la méthode Nudged Elastic Band (NEB). L’un des principaux défis de cette méthode est qu’elle est coûteuse en termes de calcul. Il y a deux raisons principales pour cela. La première est qu’il faut un grand nombre d’images pour augmenter la résolution de la recherche. L'autre raison est que le principe de recherche n'est pas variationnel (c'est-à-dire minimiser une fonction objectif), donc le nombre de mises à jour par image a également tendance à être important.
La méthode nouvellement mise en œuvre dans cette étude résout ces problèmes de manière innovante. Premièrement, le nombre d’images peut être réduit à environ 3, puisque seule la région autour de l’état de transition est recherchée de manière intensive. De plus, le principe de recherche est variationnel, ce qui permet de le résoudre plus efficacement. Plus précisément, la fonction objectif est définie comme l'intégrale linéaire de l'exponentielle de l'énergie le long du trajet.
Les performances de la nouvelle méthode ont été évaluées sur 121 réactions chimiques et les résultats ont été comparés à la méthode NEB et sa version améliorée. Premièrement, la méthode actuelle a correctement identifié les états de transition dans 98 % des cas. Cette précision est bien supérieure à celle de la méthode NEB et comparable à la version améliorée. Deuxièmement, la méthode actuelle a montré une réduction significative du coût total de calcul :environ 70 % de moins que la méthode NEB et 50 % de moins que sa version améliorée.
Pour faciliter une application plus large, les chercheurs ont rendu leur programme informatique disponible sur GitHub. Écrit en Python et conçu pour être utilisé avec l'environnement de simulation atomique (ASE), il permet aux chercheurs d'explorer facilement les états de transition en spécifiant les réactifs et les produits.
Pour l’avenir, les implications de cette recherche sont vastes. En rendant les recherches d'états de transition plus faciles et plus rapides, la méthode est sur le point d'accélérer la recherche et les développements dans tous les domaines des sciences naturelles utilisant la chimie computationnelle.
La recherche est publiée dans le Journal of Chemical Theory and Computation. .
Plus d'informations : Shin-ichi Koda et al, Localisation des états de transition par optimisation variationnelle du chemin de réaction avec une fonction objective sans dérivé énergétique, Journal of Chemical Theory and Computation (2024). DOI :10.1021/acs.jctc.3c01246
Fourni par les Instituts nationaux des sciences naturelles