Dans un article récemment publié dans Nature Communications , le groupe de recherche en modélisation des protéines HUN-REN-ELTE (Institut de chimie) a posé les bases d'une méthode mathématique permettant la comparaison assistée par ordinateur des structures tridimensionnelles des protéines. La méthode est unique dans la mesure où, alors que les alternatives disponibles jusqu'à présent ne prenaient en compte que la position des atomes, la nouvelle technique, appelée LoCoHD (Local Composition Hellinger Distance), inclut également les informations chimiques des atomes.
Les protéines sont des machines moléculaires qui exécutent les processus nécessaires au fonctionnement des cellules, agissant comme des commutateurs moléculaires, transcrivant les informations de l'ADN, transportant de petites et grandes molécules et régulant les réactions chimiques liées au métabolisme. Cependant, pour que tout cela réussisse, la protéine en question doit avoir la bonne conformation spatiale, c'est-à-dire son propre arrangement 3D correct.
Plusieurs méthodes expérimentales (cristallographie aux rayons X, spectroscopie de résonance magnétique nucléaire, cryomicroscopie électronique) sont disponibles pour déterminer la disposition des atomes dans une protéine et, au cours des dernières décennies, les chercheurs en protéines ont découvert la forme de près de 220 000 protéines. Ces résultats nécessitent de plus en plus le développement de méthodes informatiques capables d'analyser ces arrangements.
L'une de ces méthodes est l'algorithme appelé LoCoHD, développé par Zsolt Fazekas, titulaire d'un doctorat. candidat à l'école de chimie ELTE Hevesy György et chercheur dans le groupe de recherche du Dr András Perczel. L'algorithme compare les environnements locaux autour des acides aminés dans les protéines en fonction de leur nature chimique (par exemple, composition élémentaire, charge, hydrophobicité, etc.).
La méthode décide sur une échelle simple de 0 à 1 dans quelle mesure les structures en question sont différentes les unes des autres. Des valeurs proches de 0 suggèrent une grande similarité entre les arrangements atomiques et les propriétés chimiques, tandis que des valeurs proches de 1 indiquent que les protéines comparées peuvent avoir des propriétés très différentes. La valeur numérique résultante (appelée métrique) peut ainsi être utilisée pour obtenir de nouvelles informations sur le système étudié.
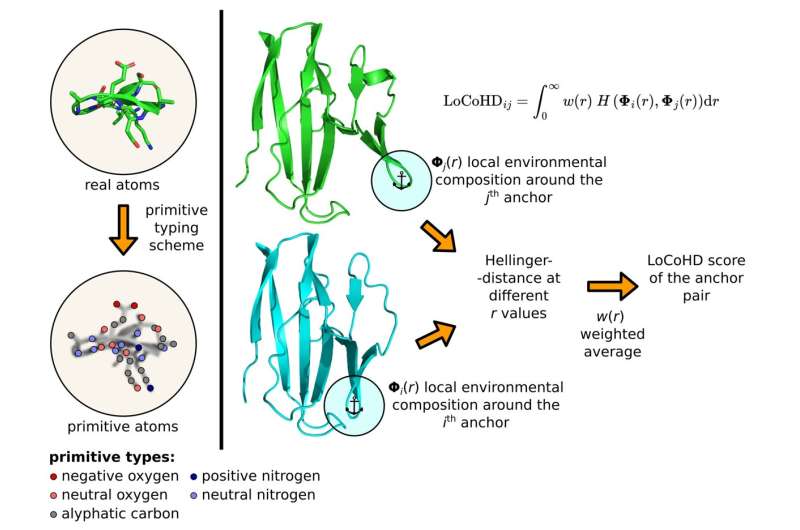
L'algorithme utilise un protocole en plusieurs étapes pour générer le nombre représentant les différences structurelles. Dans un premier temps, il convertit les atomes réels de la protéine en atomes dits primitifs. Ceux-ci peuvent être représentés sous forme de positions virtuellement étiquetées dont les étiquettes indiquent la nature chimique de l'atome d'origine.
Ainsi, par exemple, un atome primitif peut être un « azote chargé positivement », un « oxygène chargé négativement », un « oxygène chargé neutre », un « carbone aromatique », etc. Les étiquettes sont générées selon un système dit primitif. schéma de typage, qui nous indique sous forme de tableau comment convertir des atomes réels en atomes primitifs. L'utilisateur peut librement préciser ce tableau, fixant la résolution chimique de la méthode.
La deuxième étape consiste à déterminer les points de référence de la comparaison en sélectionnant un sous-ensemble d’atomes primitifs. Ces atomes primitifs spéciaux sélectionnés sont appelés atomes d’ancrage. Pour chaque paire d'atomes d'ancrage sélectionnée, l'algorithme effectue une étape de comparaison dont le résultat donne la mesure de dissimilarité souhaitée. Ces nombres peuvent être utilisés au niveau local, ou ils peuvent être moyennés en un seul descripteur caractérisant la protéine entière.
Dans l'étude, les chercheurs ont souligné que la méthode peut également être utilisée dans le cadre des concours semestriels CASP (Critical Assessment of Protein Structure Prediction), un concours bien connu dans le domaine de la recherche sur les protéines. Lors de cet événement, les concurrents utilisent différents algorithmes pour modéliser la forme de protéines ayant des structures encore inédites. Les juges du CASP utilisent un certain nombre de méthodes de comparaison de structure pour évaluer les candidats, mais aucune d'entre elles ne prend en compte la chimie des environnements locaux d'acides aminés.
À l’aide des données du concours CASP14 2020, les chercheurs ont effectué une analyse comparative de plusieurs protéines modélisées, y compris les structures prédites par la méthode AlphaFold2 basée sur l’intelligence artificielle. Parmi ceux-ci, ils ont mis en avant l’analyse d’une protéine du virus SARS-CoV-2 appelée ORF8. Dans les structures modélisées de cette protéine, des environnements d'acides aminés ont été identifiés qui diffèrent considérablement dans leurs modèles d'interaction des environnements trouvés dans la structure expérimentale.
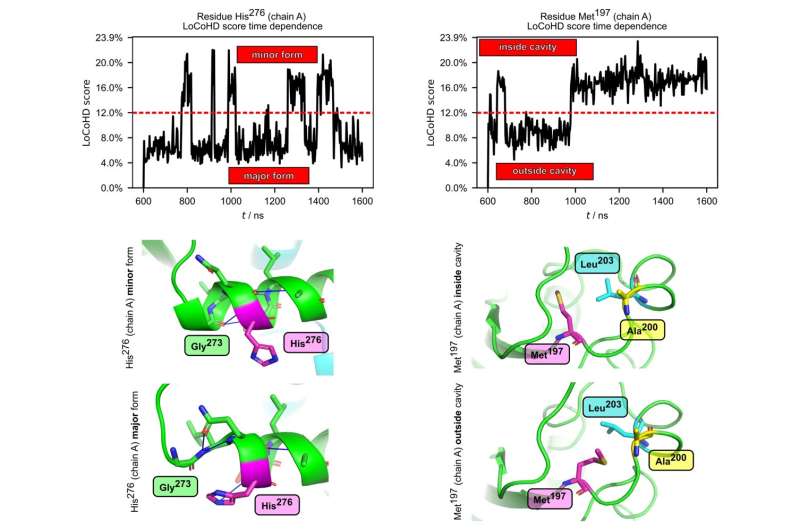
En plus d’étudier les structures statiques, les chercheurs ont également vérifié si la méthode était adaptée à l’analyse du mouvement interne des protéines. Ils ont utilisé des simulations capables de reproduire des mouvements moléculaires et des données extraites d'ensembles structurels. L'un des systèmes étudiés était la protéine podocine, qui remplit des fonctions vitales dans le rein et dont les mutations peuvent provoquer des maladies graves, souvent mortelles.
La méthode LoCoHD a été utilisée pour identifier les acides aminés de la protéine qui subissent des changements chimiques et environnementaux majeurs lors du mouvement de la podocine, pouvant affecter à la fois sa structure et sa fonction. De même, la méthode LoCoHD a été appliquée avec succès dans l'étude de la protéine capside du VIH-1, dans laquelle un acide aminé essentiel à la formation de l'enveloppe virale a été identifié.
Ces résultats ne sont pas seulement des curiosités de recherche, mais en étudiant plus efficacement les structures des protéines, nous pouvons nous rapprocher d'une meilleure compréhension des agents pathogènes responsables de maladies graves et du développement de médicaments et de thérapies efficaces.
Plus d'informations : Zsolt Fazekas et al, LoCoHD :une métrique pour comparer les environnements locaux de protéines, Nature Communications (2024). DOI :10.1038/s41467-024-48225-0
Informations sur le journal : Communications naturelles
Fourni par l'Université Eötvös Loránd